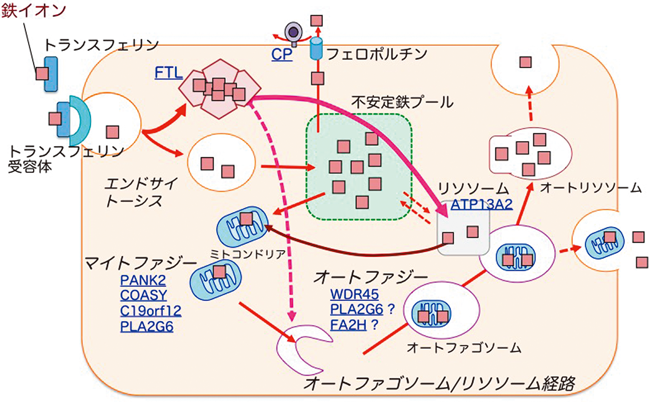
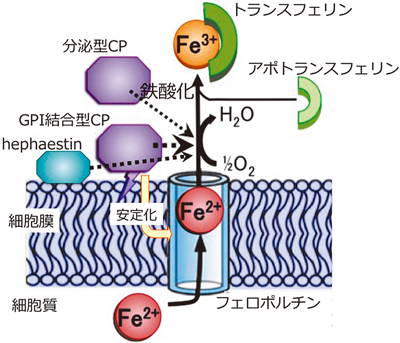
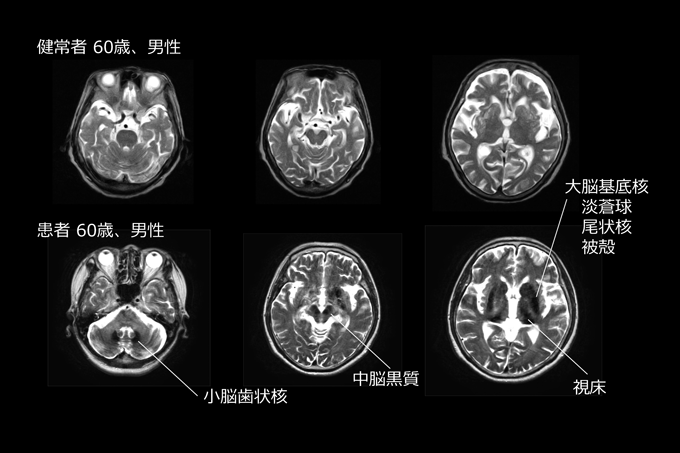
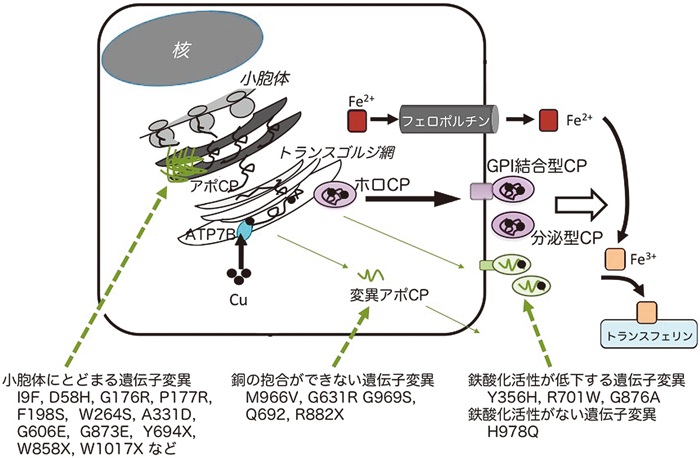
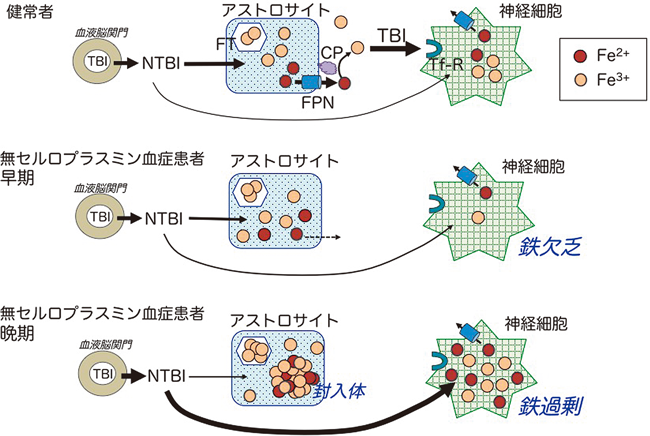
過剰な鉄蓄積による神経変性症Neurodegeneration with brain iron accumulation
浜松医科大学内科学第一講座First Department of Medicine, Hamamatsu University School of Medicine ◇ 〒431–3192 浜松市東区半田山1丁目20–1 ◇ 1–20–1 Handayama, Higashi-ku, Hamamatsu 431–3192
発行日:2018年6月25日Published: June 25, 2018