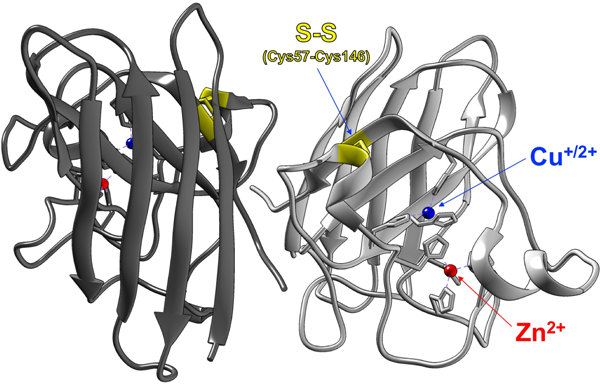
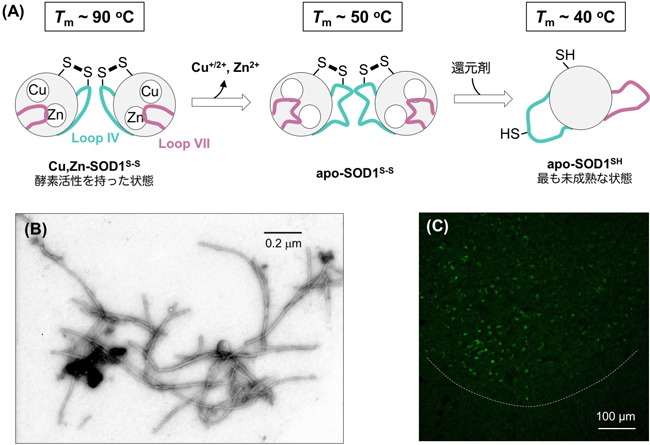
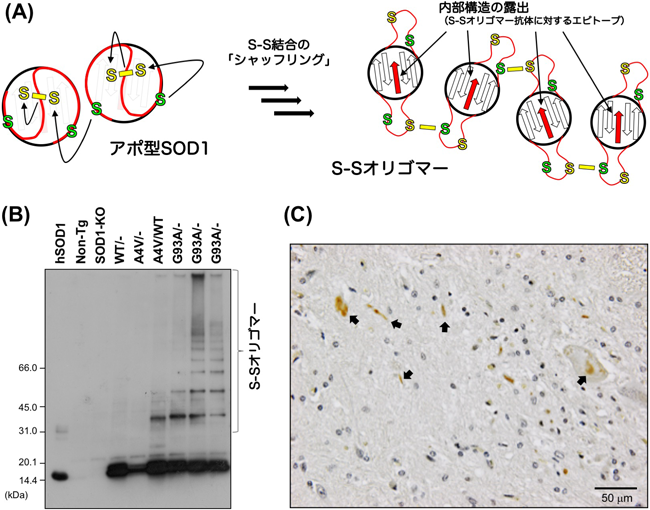
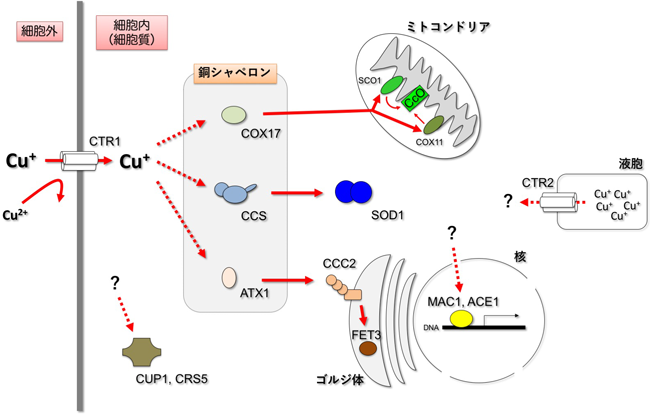
生体内銅イオン動態に着目した筋萎縮性側索硬化症の病理解明Pathological roles of copper dynamics in amyotrophic lateral sclerosis
慶應義塾大学理工学部Department of Chemistry, Keio University ◇ 〒223–8522 神奈川県横浜市港北区日吉3−14−1 ◇ 3–14–1 Hiyoshi, Kohoku, Yokohama, Kanagawa 223–8522, Japan
発行日:2018年6月25日Published: June 25, 2018