ファンコニ貧血(Fanconi anemia:FA)はスイスの小児科医Guido Fanconi博士によって1927年に報告された先天性骨髄不全症候群の1つである.X染色体上に存在するFANCBなどを除き基本的には常染色体潜性遺伝であり,100,000出生に1人程度の発症頻度を有する.その本態はFANC遺伝子群の異常によって,DNA鎖間架橋(interstrand crosslink:ICL)を中心としたDNA損傷を修復できず,細胞死やゲノム不安定性が引き起こされることにある.そのためFA患者細胞は,ICLを導入する薬剤(マイトマイシンCなど)によって高度な染色体断裂が引き起こされ,また細胞死誘導においてきわめて強い感受性を持つ.これらはFAの特徴とされている1–3).
実際にFA患者の細胞内においてICLを含めたDNA損傷をもたらしているのは,少なくとも部分的には内因性アルデヒドであると考えられている.特に,アセトアルデヒド,ホルムアルデヒド等は細胞内においてDNA間,DNA-タンパク質間などさまざまな架橋を形成することでダメージを与えることが示唆されている.アセトアルデヒドの分解は主にアルデヒドデヒドロゲナーゼ2(aldehyde dehydrogenase 2:ALDH2)によるが,FANCD2とALDH2のダブルノックアウトマウスを用いた解析では,造血幹細胞においてDNA損傷の蓄積がみられることが報告されている4).臨床的には,FA患者がアセトアルデヒドを分解できない遺伝子バリアント(ALDH2 pE487K)をホモで持つ場合,骨髄異形成症候群(myelodysplastic syndrome:MDS)/白血病への進展が著しく早まることが観察されている5, 6).これらの結果から,アルデヒドに注目したファンコニ貧血患者に対する治療応用も期待される.またFA遺伝子の異常は造血幹細胞を支持する骨髄間質細胞へも影響することも報告されており,FA遺伝子欠損の影響は直接的な造血細胞への障害だけでは説明できない可能性もある7).しかし,なぜ造血幹細胞がファンコニ貧血において特異的に損傷を受けるのかについてはまだ未解明の部分が多く,今後の検討が待たれる.
ICLとはDNA二重鎖の対合する塩基が共有結合によって物理的につながってしまうDNA損傷を指す.ICLによって,DNAの複製や転写の際に二重鎖が分離されず,反応が停止してしまうきわめて有害なDNA損傷の一つである.上記のようにFA患者細胞はICL修復に障害を持つが,逆にいえばFA患者細胞を調べることで,ICL修復に関する分子メカニズムを解き明かすことが可能になる.そのため世界各地の研究室で患者変異の解析が精力的に行われてきた.FANC遺伝子群は,FA患者での変異が同定されて初めてFANC~という遺伝子名が付与されるため,FANCAからいくつかの欠番を含んでアルファベット順に数え,現在FANCVまで,計21の原因遺伝子が同定されてきた(表1).このようにICLの原因遺伝子群の全体像はみえつつあるが8–10),それら遺伝子産物の詳細なICL修復制御機構についてはいまだに不明な点が多い.
表1 現在までに同定されているファンコニ貧血原因遺伝子| 遺伝子 | 頻度 | 関連するタンパク質 | 分子機能 |
|---|
| FANCA | 71% | FANCG | FAコア複合体 |
| FANCB | まれ | FANCL | FAコア複合体 |
| FANCC | 7% | FANCE | FAコア複合体 |
| FANCE | 4% | FANCC | FAコア複合体 |
| FANCF | 2% | FANCA/G, C/E | FAコア複合体 |
| FANCG | 13% | FANCA | FAコア複合体 |
| FANCL | まれ | UBE2T | FAコア複合体E3リガーゼ |
| FANCM | 1例のみ | FANCF, FAAP24 | FAコア複合体 |
| FANCT/UBE2T | まれ | FANCL | FAコア複合体 |
| FANCD2 | 1% | FANCI, FANCE | ID複合体 |
| FANCI | まれ | FANCD2 | ID複合体 |
| FANCP/SLX4 | まれ | XPF, FANCD2 | ヌクレアーゼのリクルート |
| FANCQ/XPF | まれ | SLX4, ERCC1 | ヌクレアーゼ |
| FANCV/REV7 | まれ | REV1, REV3 | 損傷乗り越え修復(TLS) |
| FANCD1/BRCA2 | 1% | Rad51 | 相同組換え |
| FANCJ/Brip1 | まれ | BRCA1 | 相同組換え |
| FANCN/PALB2 | まれ | BRCA1, BRCA2 | 相同組換え |
| FANCO/Rad51C | まれ | Rad51パラログ | 相同組換え |
| FANCR/RAD51 | まれ | BRCA1, BRCA2, PALB2 | 相同組換え |
| FANCS/BRCA1 | まれ | BRCA2, PALB2, RAD51 | 相同組換え |
| FANCU/XRCC2 | まれ | RAD51パラログ | 相同組換え |
| 現在までにRFWD3を含めて22の原因遺伝子が同定されている.それぞれの頻度,関連する因子およびFA経路における役割を示す. |
このような状況の中で,ドイツ人患児の全エキソン解析から,新規ファンコニ遺伝子候補としてRFWD3が同定された.この分子については,1)ユビキチン化を介してp53を安定化する11),2)Chk1のリン酸化を介した細胞周期コントロールに必要である,2)複製,修復に必須な一本鎖DNA結合タンパク質であるRPA(replication protein A)と会合する12, 13),という報告があるものの機能面では明確な結論は出ていなかった.そこで我々はRFWD3が真にファンコニ貧血の原因遺伝子FANCWであることを証明し,さらにその分子機能を解明するために研究を開始した.
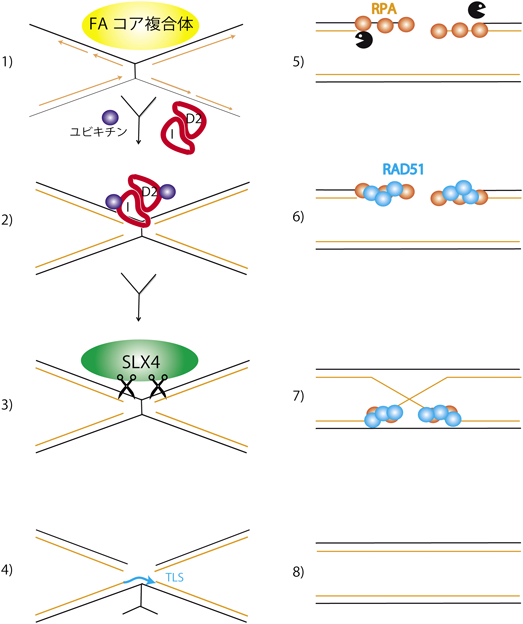
ICL修復経路は相同組換え修復(homologous recombination repair:HR)に加えてヌクレオチド除去修復(nucleotide excision repair:NER),損傷乗り越え修復(translesion synthesis:TLS)など種々のDNA修復形式を組み合わせた複雑なプロセスである(全体像を図1に示す).きわめて有害なICLを,適切に修復因子をコーディネートした上でDNA二重鎖切断(DNA double-strand break:DSB)を導入し,HRで修復すると解釈すると理解しやすい.
順を追って説明すると,1)ICL損傷部位での複製フォークの停止およびFAコア複合体の活性化,2)コア複合体によるFANCD2-FANCI複合体(ID複合体)のモノユビキチン化,3)SLX4/FANCPを中心としたヌクレアーゼ複合体のリクルート,4)ヌクレアーゼによるICL両側の切断(unhookingと呼ばれるプロセス)によるDSB導入,および損傷乗り越え修復(TLS),5)DNA平滑末端の削り込みによる一本鎖DNA(single-strand DNA:ssDNA)の生成およびRPAによる結合,6)BRCA1/2等によるRAD51のssDNAへのローディング(RAD51 filament形成),7)鋳型鎖への侵入およびDNA合成,8)修復の完了,という過程で行われると考えられている.このうち,key switchとなるのが2)ID複合体のモノユビキチン化であり,FANC遺伝子群の八つがこのプロセスに必要なFAコア複合体の構成成分をコードしている(表1).
HRは5)~8)のDSB修復過程に相当する.HRは姉妹染色分体を鋳型として必要とし,S期,G2期に主に機能するerror-freeな修復様式であり,細胞周期を通じて行われるerror-proneな非相同末端結合(non-homologous end joining:NHEJ)と対照的である.HR関連遺伝子にはFAの原因となるものが多数あり,現在までにBRCA2, BRIP1, PALB2, RAD51Cなどが原因遺伝子として同定されてきた14–16).さらに最近BRCA1およびRAD51, XRCC2など複数の遺伝子が加わり17, 18),今後もHR関連遺伝子はFA原因遺伝子として追加されていくものと予想される.また,BRCA1, BRCA2, PALB2, BRIP1などに関しては片アレル変異で家族性乳がんが発症し,両アレルの変異ではファンコニ貧血を発症するか,変異の重症度によっては致死になると考えられており,残存する活性によって表現型が異なってくるものと思われる.
4. 新規ファンコニ遺伝子RFWD3/FANCWの同定
臨床面からみると,ファンコニ貧血患児では皮膚の色素沈着,身体奇形,低身長,性腺機能不全などを伴うが,その表現型は実に多彩である.最も重大な合併症は,小児期に発症する骨髄不全であり,思春期から成人期にかけて骨髄異形成症候群(MDS)や急性骨髄性白血病への移行がみられることが多いため,しばしば骨髄移植による治療を要する19, 20).
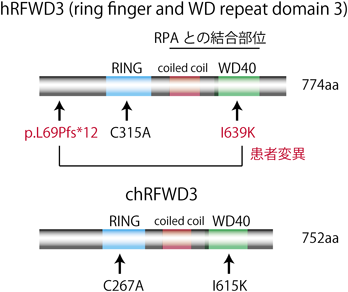
本例(RFWD3に複合ヘテロ変異を持つ患児)は低出生体重児であり(妊娠35週で1850 g),十二指腸閉鎖,両側母指欠損,小頭症,キアリ奇形1型,下垂体機能不全などさまざまな身体奇形および臓器不全を合併していたが,知能に問題はなく,12歳時点では通常の学校に通学している.骨髄では血球産生の低下が認められ軽度の異形成が観察されるものの,末梢血中では特に血球減少は認めていない.このようなケースでは身体症状からの診断は難しいが,末梢血リンパ球におけるマイトマイシンC(MMC)添加による染色体断裂試験が有用である.ファンコニ貧血の患者細胞では,ICL導入薬剤であるMMCによって高度な染色体断裂像がみられることが佐々木正夫博士(京都大学放射線生物研究センター前教授)によって発見され,本試験は現在もファンコニ貧血診断のゴールドスタンダードとして用いられている21).本例では,患者の末梢血リンパ球および繊維芽細胞を用いた検証でMMCによる高度な染色体断裂が惹起され,そのレベルはRFWD3相補にて抑制されたため,FAとして矛盾しないことが確認された22).全エキソン解析で同定された本患者は,RFWD3遺伝子にトランケーションを伴うフレームシフト変異(c.205_206dupCC;p.L69Pfs*12)と,保存領域における点突然変異(c.1916T>A;p.I639K)の複合ヘテロ変異を有していたが(図2),前者はタンパク質の発現が認められず,後者はタンパク質の発現はあるが機能不全を来す変異であることが推定された16).
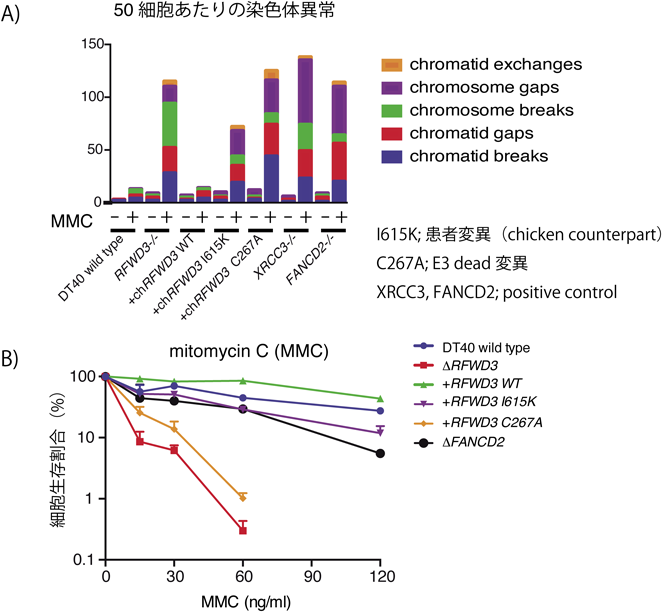
RFWD3変異が真にファンコニ貧血の原因であることを確認するため,まず,我々はDT40細胞(ニワトリB細胞腫瘍株)においてRFWD3ノックアウト細胞を作製し,ファンコニ貧血の表現型が認められるか検証した.DT40 ΔRFWD3では,患者細胞と同様,MMCによって高度な染色体断裂が惹起され(図3A),またMMCによって細胞死が誘導され,強い感受性を持っていた(図3B).また,この細胞に野生型のRFWD3を再度発現させることで,MMCに対する染色体脆弱性と生細胞率低下をキャンセルすることができた.さらに,ノックアウトマウス由来の胎仔繊維芽細胞を用いた実験でも同様の結果が得られた.以上より,RFWD3がファンコニ貧血を引き起こす責任遺伝子であることは確実と考えられた.
次に,我々は患者変異がRFWD3の分子機能にどのような影響をもたらしているかを検証した.RFWD3は図2に示すように,774アミノ酸からなるRING型のE3リガーゼである.患児は複合ヘテロ変異を有していたが,トランケーションを引き起こす変異アレルについてはタンパク質としての発現を認めず,明らかにヌル変異である.一方,hRFWD3 I639K(ニワトリではI615Kに相当)のミスセンス変異(WD40変異)について検証が必要であった.
まず,DT40 ΔRFWD細胞にさまざまな変異を有したニワトリ(ch)RFWD3を発現させたものを比べると,1)WD40変異体ではMMCによる染色体断裂を部分的にしか抑制できないこと,2)RINGドメイン変異体(chRFWD3 C267A,図2参照)を発現した細胞はΔRFWD3細胞とほぼ同様の表現型を示すことが観察された(図3A, B).RINGドメインの変異によってE3酵素としての活性が消失するため,ICL修復にはRFWD3のユビキチン化活性が必要であり,WD40変異体は部分的な機能障害を有することが示唆された.
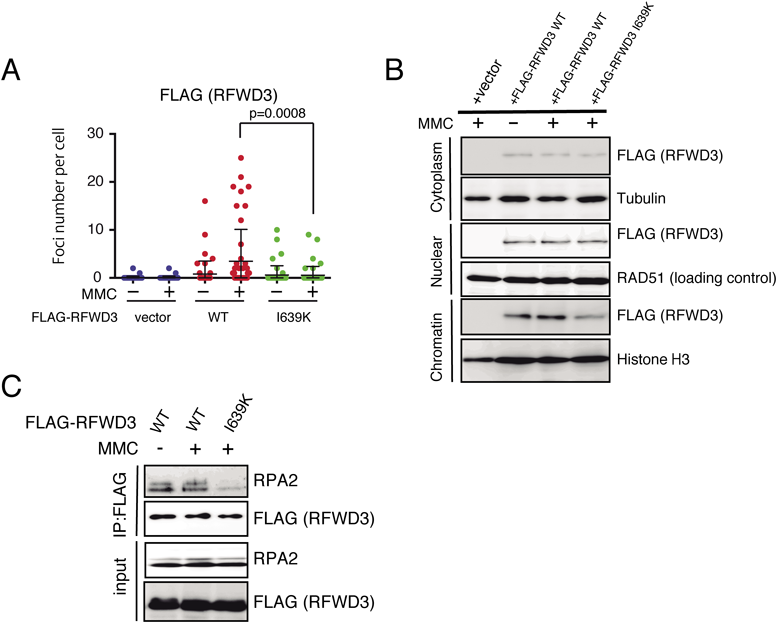
次に,WD40変異体における機能不全のメカニズムを検証するため,変異タンパク質の細胞内局在を検証した.U2OS細胞に野生型hRFWD3とWD40ドメインの変異体I639K hRFWD3を発現させてMMC刺激下での局在を免疫染色で評価したところ,後者ではDNA損傷後のフォーカス形成(DNA修復タンパク質は,一般にDNA損傷部位に集積するので,免疫染色でドット状の分布を示す)が非常に低下していることが明らかになった(図4A).さらに,野生型hRFWD3はクロマチン画分にほとんど検出されず(図4B),このWD40ドメインの変異は局在異常をもたらす病原性の変異であると考えられた.
では,なぜこの変異がクロマチンへの集積を低下させるのであろうか.既報ではRFWD3はRPAと結合してフォーカス形成することが報告されており,I639K変異はタンパク質間の会合に関与するWD40ドメインにある.このことから,I639K変異はRPAとの結合に影響するという仮説が示唆され,免疫沈降では予想どおりI639K変異は野生型に比べてRPAとの結合低下を示した(図4C).一方で,我々は免疫沈降によってRFWD3が二量体あるいは多量体を形成することを見いだしたが,I639K変異は二量体形成には影響しなかった.
これらの結果より,患児の複合ヘテロ変異は,片方のアレルからは発現せず,もう片方のアレルから発現するタンパク質は局在異常による機能不全があり,結果としてファンコニ貧血の原因となっていることが示された.
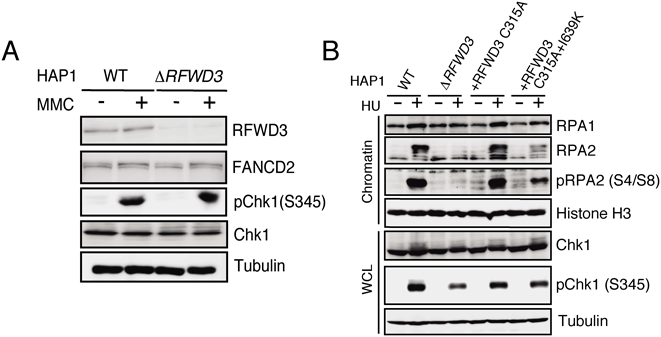
次に,RFWD3がどのような分子機能を果たしているかを,ヒト細胞株HAP1においてRFWD3ノックアウト細胞を作製して調べることにした.FA経路は大きくFANCD2のモノユビキチン化の上流と下流に分けられる(図1).HAP1 ΔRFWD3では,MMC刺激によってFANCD2が正常にモノユビキチン化されることから,RFWD3の作用点はそれより下流にあると考えられた.そこで,RFWD3のHRにおける機能を検討したところ,DT40 ΔRFWD3ではI-SceI発現によるHRレポーターアッセイ(制限酵素であるI-SceIによる切断後,HRによって不完全な遺伝子が修復されて発現するようになることを利用したアッセイ系)と遺伝子ターゲティング効率が顕著に低下しており,強いHR欠損が示唆された.
では,RFWD3はどのようにHRを制御しているのであろうか.まず我々は,HRの必須因子であるRPAとRAD51の細胞内分布がΔRFWD3細胞でどう変化するかを検証した.すると,ΔRFWD3細胞ではRPAとRAD51がMMC刺激下でクロマチンに多く残留し,かつRPAとRAD51フォーカスの共局在する割合が増加していた.この共局在という現象は,抗RPA2抗体と抗RAD51抗体を用いたPLAアッセイ(proximity ligation assay,細胞内のタンパクの相互作用を可視化する方法の一つ.2種の抗体のFc部分に相補的なオリゴヌクレオチドを結合させ,ローリングサークル増幅を介して,互いの標的が近接している時のみシグナルが得られるようにデザインされている)でも確認された.PLAの空間分解能は40 nmといわれており,同じ一本鎖DNA分子上にRPAとRAD51が混在している可能性が考えられる.
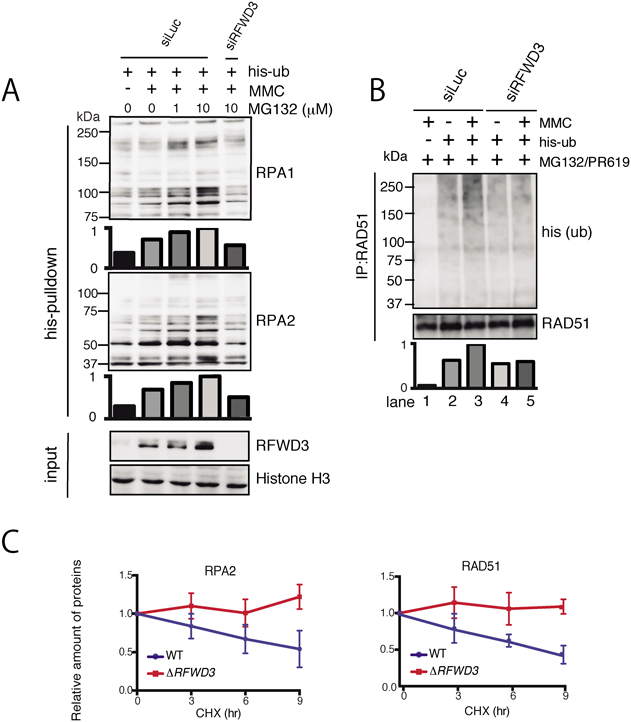
ここで,RFWD3の機能を考えるにあたってその構造に立ち返ってみたい.RFWD3はRING型E3リガーゼであり,ユビキチンを基質タンパク質に付加する酵素活性を持つ.したがって,基質を同定することがその機能を知るための第一歩となる.我々は,ΔRFWD3細胞でRPAとRAD51がDNA損傷後のクロマチン上に野生型細胞に比較してより多く存在するという現象から,両者がRFWD3の基質である可能性を考えた.そこで,細胞内にユビキチンを強制発現させ感度を高めて検討したところ,RFWD3は効率よくRPAとRAD51をユビキチン化することがわかった23).RFWD3と対応する可能性のあるE2酵素群を精製し,RPAやRAD51とまぜて反応させたところ,in vitroでのユビキチン化を検出することもできた.興味深いことに,in vivoではユビキチン化はMMCによるDNA損傷刺激に依存し,プロテアソーム阻害薬であるMG132によってユビキチン化したRPAおよびRAD51が蓄積した(図5A, B).したがって,RFWD3はこれらのタンパク質をDNA修復経路の中で分解に導いている可能性が示唆された.
そこで,シクロヘキシミド(CHX)でタンパク質合成を阻害し,細胞内のRPAとRAD51量をウェスタンブロットによって定量し,その分解速度を検証した.HAP1由来ΔRFWD3細胞では,野生型細胞(WT)に比べてMMCによるDNA損傷刺激後,両者の減少速度が低下しており,RFWD3によるユビキチン化がプロテアソームによる分解につながっていることが支持された(図5C).
上述の結果から,RFWD3のHRにおける主要な役割は,RPAとRAD51をユビキチン化依存性にクロマチンから除去し,プロテアソームによる分解に導くことにあると考えられた.では,ユビキチン化がどのようにしてこれらのタンパク質のクロマチンからの除去を促進しているのであろうか.
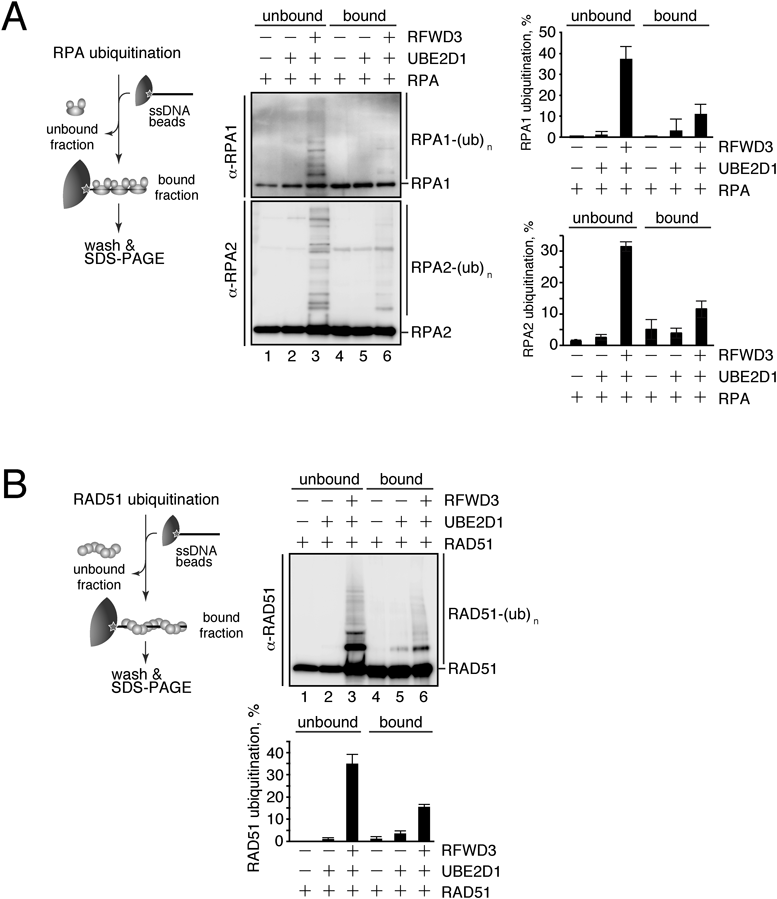
そこで,我々はまず,ユビキチン化によってRPAがssDNAへの親和性を変化させる可能性を検証した.in vitroでユビキチン化したRPAとssDNAビーズを混和し,ビーズに結合したRPAと非結合のRPAを比較したところ,興味深いことにユビキチン化されたRPAは多くが非結合の画分に認められた(図6A).このことは,RPAはユビキチン化されるとssDNAヘの親和性が著しく低下し,解離しやすくなることを示唆する.同様の実験をRAD51に対して行うと,やはりユビキチン化されることでssDNAへの親和性が低下するという結果が得られた(図6B).
では,クロマチンへの結合が低下したPRAとRAD51はどのようにプロテアソームによって分解されるのか.直接的にプロテアソームがクロマチンにアクセスする可能性も考えられるが,さまざまなタンパク質がユビキチン化による制御を受けている場所にプロテアソームが近接する状況は細胞にとって危険性が高く,輸送タンパク質による介在が合理的と考えられる.そこで,我々は,valosin-containing protein(VCP)/p97に注目した.VCPはATP依存性にユビキチン化されたタンパク質を認識して細胞内の構造体から引き離しプロテアソームに輸送するシャペロンタンパク質であり,近年DNA修復を含め,さまざまな細胞内プロセスへの関与が報告されている24, 25).RPAおよびRAD51もユビキチン化されるので,VCPの標的タンパク質である可能性を考えた.そこで共免疫沈降で検証したところ,MMCによるDNA損傷とRFWD3依存性に,RPAとRAD51の両方がVCPと会合することが確認できた.
細胞内のタンパク質の挙動を解析する方法として,fluorescent recovery after photobleaching(FRAP)がある.FRAPでは細胞内の一部に局在する蛍光タンパク質に強いレーザー励起光を照射して退色させ,照射野外からの流入による蛍光の回復を定量的に測定する.クロマチン上に局在した蛍光タグつきのタンパク質にFRAPを行うことで,その入れ替わりのダイナミクスを定量化することができる.我々はGFP-RPA2やRAD51-GFPを発現させたU2OS細胞を用いて上記の結果を検証した.ΔRFWD3細胞では,クロマチン上に存在しているRPAおよびRAD51(フォーカスとしてドット状にみえる)がユビキチン化されず除去されにくいため,タンパク質のターンオーバーが低下し,FRAPではシグナルの回復が低下すると推測される.結果は予想どおりで,RFWD3をsiRNAによってノックダウンするとGFP-RPA2, RAD51-GFPの双方に関して,蛍光退色後のシグナルの回復が低下した.重要なことに,VCPを除去した細胞でも同じ結果が得られたため,VCPはこれらのタンパクのターンオーバーに必要であることが示唆された.
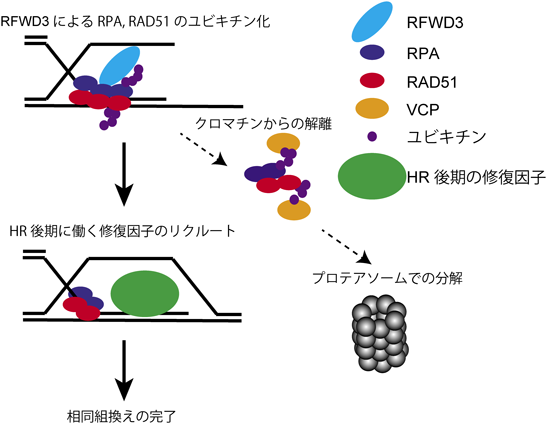
上記の結果をまとめると,RFWD3によるRPAとRAD51のユビキチン化はクロマチンからの解離を促し,また,RPAとRAD51はVCPに介在されてプロテアソームに輸送され分解につながっていくと考えられる.
RPAとRAD51はともにHRの必須因子であることから,RFWD3によるユビキチン化によって不適切なタイミングで分解されると,HRが抑制されるはずであり,RFWD3によるRPAとRAD51の分解は厳密に制御されている必要がある.では,RFWD3の活性はどのように制御されているのであろうか.DNA損傷応答においては,しばしばリン酸化がトリガーとなるが,RFWD3のS46/S63の二つの残基がATRおよびATMの双方によってリン酸化されることがすでに報告されていた11).そこで,DNA損傷後に活性化される三つのキナーゼ,ATM, ATR, DNA-PKの阻害剤を用いて,RFWD3のリン酸化を検討したところ,ATMとATRを同時に阻害すると有意な低下が観察された.我々はこのセリンをアラニンに置換し,リン酸化されない変異体(RFWD3 S46/63A)を作製し,その影響を検証した.まず,RFWD3 S46/63Aを細胞に発現させてRPAのユビキチン化能をみたところ,WTに比して低下していることがわかった.また,HAP1 ΔRFWD3細胞にRFWD3 S46/63Aを発現させたところ,MMC刺激後,WT RFWD3を発現させた細胞に比して低い生存率を示し,この部位のリン酸化がRFWD3の活性化に重要であることが示唆された.しかし,RFWD3 S46/63A変異体は部分的には機能が残存しており,他のリン酸化部位など未解明の制御機構が存在していることが予想され,今後の検討課題である.
9. 残存するRPAとRAD51はDNA修復の進行を阻害する
次に我々は,なぜRFWD3によるRPAとRAD51の除去がHRの進行に必要であるのかを考えた.最も単純な仮説は,RPAとRAD51が残っていると,修復反応が進行中の局所DNAがRPAとRAD51に覆われているため後続の修復因子がアクセスできない,というものである.HR後期過程においてどのような分子が作用するのかについては,よくわかっていない部分が多いが,我々は,MCM8とRAD54をHR後期過程に関わる分子の候補として,検討を行った.
MCM8とRAD54はともにDNA損傷によってクロマチンにリクルートされることがわかっている26, 27).そこで,HAP1 ΔRFWD3細胞における影響を検証したところ,WTに比べてMMC刺激下におけるMCM8とRAD54のクロマチンへのリクルートが有意に低下していた.また,前述のようにRFWD3はVCPと協調して働くが,VCPをsiRNAでノックダウンした細胞でも,同様にMCM8やRAD54がクロマチンにリクルートされにくいことが確認できた.
次に,RPA2とRAD51がRFWD3のHRにおける主要なターゲットであるか検証するため,これらタンパク質のユビキチン化候補部位の5か所のリシンをアルギニンに置換した変異体を作製し,内在性タンパク質と入れ替える実験を行った(FLAG-RPA2 5KR, FLAG-RAD51 5KR).すると,これらの変異体はMMCによる刺激後,WTに比してクロマチン上に長く残留し,MCM8およびRAD54のクロマチンローディングが低下しているなど,RFWD3欠損と同様の表現型を示すことが明らかになった.
上記より,RPAやRAD51が除去されずにクロマチン上に残留することで,正常な修復タンパク質との入れ替わりが阻害され,HRが完了に至らないというメカニズムの存在が示唆された.
10. RFWD3はユビキチン化を介さずチェックポイント機構を活性化する
既報では,RFWD3はヒドロキシ尿素(HU)処理後のChk1のリン酸化に必要であるとされている28).しかし,一般にはRPAに会合するATRIP分子を介したATRの局所リクルートを開始点としてChk1のリン酸化が引き起こされると考えられているため,我々が見いだしたRFWD3によるRPA制御機構ではHU誘導性Chk1リン酸化への影響を説明できない.この点を検証するため,HAP1 ΔRFWD3細胞をHUとMMCで処理して比較したところ,HU処理後ΔRFWD3細胞では明らかなChk1リン酸化の減弱が認められたのに対して,MMC処理ではRFWD3欠損の影響が認められなかった(図7A).さらに,HU刺激下でのChk1リン酸化は,RFWD3を再度発現させることで回復したが,興味深いことにユビキチン化能を持たないC315A RINGドメイン変異体を発現させても回復した(図7B).このChk1のリン酸化低下はクロマチン上のRPA量の低下を伴っていた.これらの結果から,RFWD3はHUなどより長いssDNA露出が生まれる状況ではユビキチン化非依存的にRPAをクロマチン上にローディングさせるが,ICL修復過程のようにDNAの末端が削り込まれてのちのHR後期過程では,むしろユビキチン化を介したRPAの除去に働くという二面性が示唆された.
本稿では,RFWD3の新規ファンコニ貧血原因遺伝子としての発見と,HRにおける役割についての解析結果について概説した.全体像を図8に示す.RFWD3はHRを制御する必須制御因子の一つである.しかし,その活性制御についてはまだ理解不十分な部分を残しており,今後の研究によって解明する必要がある.また,これまでDNA損傷局所で一本鎖DNAに結合したRPAは,BRCA2やDSS1の働きによってRAD51と入れ替わると一般的には考えられてきたが29, 30),我々の得た知見はその概念と必ずしも一致しないように思われる.実際に細胞内でRPAとRAD51の局所集積がどのように制御されているのか,タイムコース等のより詳細な検討が望まれる.臨床面に目を向けると,RFWD3は,BRCA1やBRCA2と同様,HRに関連する因子であるため,理論的には片アレルの欠損で家族性乳がん卵巣がん症候群(hereditary breast and ovarian cancer syndrome:HBOC)の原因となる可能性があり,RFWD3の変異もBRCA遺伝子が正常な患者では検索の対象とする必要性が考えられる.HBOCについては病態および治療法についての研究が精力的に進められている.今後の研究成果を待ちたい.
謝辞Acknowledgments
この研究は,ドイツのDetlev Schindler氏との共同研究にはじまり,多くの共同研究者の方々のご協力により可能となりました.特に,早稲田大学(現東京大学)の胡桃坂仁志教授,佐藤浩一博士の生化学的解析の貢献は多大なものがあります.共同研究者のみなさまに深甚なる感謝の意を表します.
利益相反
全著者ともに,申告すべき利益相反(conflicts of interests)状態はない.
引用文献References
1) Kottemann, M.C. & Smogorzewska, A. (2013) Nature, 493, 356–363.
2) Schneider, M., Chandler, K., Tischkowitz, M., & Meyer, S. (2015) Clin. Genet., 88, 13–24.
3) Schwartz, R.S. & D’Andrea, A.D. (2010) N. Engl. J. Med., 362, 1909–1919.
4) Garaycoechea, J.I., Crossan, G.P., Langevin, F., Daly, M., Arends, M.J., & Patel, K.J. (2012) Nature, 489, 571–575.
5) Hira, A., Yabe, H., Yoshida, K., Okuno, Y., Shiraishi, Y., Chiba, K., Tanaka, H., Miyano, S., Nakamura, J., Kojima, S., Ogawa, S., Matsuo, K., Takata, M., & Yabe, M. (2013) Blood, 122, 3206–3209.
6) Garaycoechea, J.I. & Patel, K.J. (2014) Blood, 123, 26–34.
7) Zhou, Y., He, Y., Xing, W., Zhang, P., Shi, H., Chen, S., Shi, J., Bai, J., Rhodes, S.D., Zhang, F., Yuan, J., Yang, X., Zhu, X., Li, Y., Hanenberg, H., Xu, M., Robertson, K.A., Yuan, W., Nalepa, G., Cheng, T., Clapp, D.W., & Yang, F.C. (2017) Haematologica, 102, 1017–1027.
8) van Twest, S., Murphy, V.J., Hodson, C., Tan, W., Swuec, P., O’Rourke, J.J., Heierhorst, J., Crismani, W., & Deans, A.J. (2017) Mol. Cell, 65, 247–259.
9) Chen, X., Bosques, L., Sung, P., & Kupfer, G.M. (2016) Oncogene, 35, 22–34.
10) Sobeck, A., Stone, S., & Hoatlin, M.E. (2007) Mol. Cell. Biol., 27, 4283–4292.
11) Fu, X., Yucer, N., Liu, S., Li, M., Yi, P., Mu, J.J., Yang, T., Chu, J., Jung, S.Y., O’Malley, B.W., Gu, W., Qin, J., & Wang, Y. (2010) Proc. Natl. Acad. Sci. USA, 107, 4579–4584.
12) Gong, Z. & Chen, J. (2011) J. Biol. Chem., 286, 22308–22313.
13) Liu, S., Chu, J., Yucer, N., Leng, M., Wang, S.Y., Chen, B.P., Hittelman, W.N., & Wang, Y. (2011) J. Biol. Chem., 286, 22314–22322.
14) Vaz, F., Hanenberg, H., Schuster, B., Barker, K., Wiek, C., Erven, V., Neveling, K., Endt, D., Kesterton, I., Autore, F., Fraternali, F., Freund, M., Hartmann, L., Grimwade, D., Roberts, R.G., Schaal, H., Mohammed, S., Rahman, N., Schindler, D., & Mathew, C.G. (2010) Nat. Genet., 42, 406–409.
15) Reid, S., Schindler, D., Hanenberg, H., Barker, K., Hanks, S., Kalb, R., Neveling, K., Kelly, P., Seal, S., Freund, M., Wurm, M., Batish, S.D., Lach, F.P., Yetgin, S., Neitzel, H., Ariffin, H., Tischkowitz, M., Mathew, C.G., Auerbach, A.D., & Rahman, N. (2007) Nat. Genet., 39, 162–164.
16) Levitus, M., Waisfisz, Q., Godthelp, B.C., de Vries, Y., Hussain, S., Wiegant, W.W., Elghalbzouri-Maghrani, E., Steltenpool, J., Rooimans, M.A., Pals, G., Arwert, F., Mathew, C.G., Zdzienicka, M.Z., Hiom, K., De Winter, J.P., & Joenje, H. (2005) Nat. Genet., 37, 934–935.
17) Sawyer, S.L., Tian, L., Kähkönen, M., Schwartzentruber, J., Kircher, M., Majewski, J., Dyment, D.A., Innes, A.M., Boycott, K.M., Moreau, L.A., Moilanen, J.S., & Greenberg, R.A.; University of Washington Centre for Mendelian Genomics; FORGE Canada Consortium. (2014) Cancer Discov., 5, 135–142.
18) Wang, A.T., Kim, T., Wagner, J.E., Conti, B.A., Lach, F.P., Huang, A.L., Molina, H., Sanborn, E.M., Zierhut, H., Cornes, B.K., Abhyankar, A., Sougnez, C., Gabriel, S.B., Auerbach, A.D., Kowalczykowski, S.C., & Smogorzewska, A. (2015) Mol. Cell, 59, 478–490.
19) Kitao, H. & Takata, M. (2011) Int. J. Hematol., 93, 492–497.
20) Malric, A., Defachelles, A.S., Leblanc, T., Lescoeur, B., Lacour, B., Peuchmaur, M., Maurage, C.A., Pierron, G., Guillemot, D., d’Enghien, C.D., Soulier, J., Stoppa-Lyonnet, D., & Bourdeaut, F. (2015) Pediatr. Blood Cancer, 62, 463–470.
21) Sasaki, M.S. (1975) Nature, 257, 501–503.
22) Knies, K., Inano, S., Ramírez, M.J., Ishiai, M., Surrallés, J., Takata, M., & Schindler, D. (2017) J. Clin. Invest., 127, 3013–3027.
23) Inano, S., Sato, K., Katsuki, Y., Kobayashi, W., Tanaka, H., Nakajima, K., Nakada, S., Miyoshi, H., Knies, K., Takaori-Kondo, A., Schindler, D., Ishiai, M., Kurumizaka, H., & Takata, M. (2017) Mol. Cell, 66, 622–634.e8.
24) Meyer, H., Bug, M., & Bremer, S. (2012) Nat. Cell Biol., 14, 117–123.
25) Fujita, K., Nakamura, Y., Oka, T., Ito, H., Tamura, T., Tagawa, K., Sasabe, T., Katsuta, A., Motoki, K., Shiwaku, H., Sone, M., Yoshida, C., Katsuno, M., Eishi, Y., Murata, M., Taylor, J.P., Wanker, E.E., Kono, K., Tashiro, S., Sobue, G., La Spada, A.R., & Okazawa, H. (2013) Nat. Commun., 4, 1816.
26) Nishimura, K., Nakamura, Y., Oka, T., Ito, H., Tamura, T., Tagawa, K., Sasabe, T., Katsuta, A., Motoki, K., Shiwaku, H., Sone, M., Yoshida, C., Katsuno, M., Eishi, Y., Murata, M., Taylor, J.P., Wanker, E.E., Kono, K., Tashiro, S., Sobue, G., La Spada, A.R., & Okazawa, H. (2012) Mol. Cell, 47, 511–522.
27) Solinger, J.A., Kiianitsa, K., & Heyer, W.-D. (2002) Mol. Cell, 10, 1175–1188.
28) Elia, A.E., Wang, D.C., Willis, N.A., Boardman, A.P., Hajdu, I., Adeyemi, R.O., Lowry, E., Gygi, S.P., Scully, R., & Elledge, S.J. (2015) Mol. Cell, 60, 280–293.
29) Liu, J., Doty, T., Gibson, B., & Heyer, W.-D. (2010) Nat. Struct. Mol. Biol., 17, 1260–1262.
30) Zhao, W., Vaithiyalingam, S., San Filippo, J., Maranon, D.G., Jimenez-Sainz, J., Fontenay, G.V., Kwon, Y., Leung, S.G., Lu, L., Jensen, R.B., Chazin, W.J., Wiese, C., & Sung, P. (2015) Mol. Cell, 59, 176–187.
著者紹介Author Profile
 稲野 将二郎(いなの しょうじろう)
稲野 将二郎(いなの しょうじろう)関西電力病院血液内科 兼 関西電力医学研究所研究員.医学博士.
略歴2008年京都大学医学部を卒業後,同年より田附興風会北野病院にて初期研修,10年より血液内科後期研修医として勤務.13年より大学院生として京都大学放射線生物研究センターにてDNA修復の研究に従事する.17年6月より現職.
研究テーマと抱負現在は多発性骨髄腫をテーマに,臨床的な視点から治療に結びつく研究を開始している.今後悪性腫瘍の臨床の発展に少しでも寄与できるように努力していきたい.