細胞内の機能単位であるタンパク質は,細胞内の特定の「場所」で,特定の「タイミング」で最適な機能を発現する.よって,細胞内のタンパク質の真の機能を理解するためには,それが機能する「現場」である細胞の中で機能解析を行う必要がある.また,疾患の治療に用いられている低分子化合物やバイオ医薬品の多くは細胞内タンパク質・核酸分子を標的としているが,それらの効能(活性)も,実際効果を発揮する「細胞内」で調べることが必要不可欠となってきている.しかも,これら薬剤やバイオ医薬品は,正常な細胞内環境では無害であり,病態細胞内環境でだけその機能を発揮することが強く求められてもいる.このように,タンパク質やそれと相互作用するさまざまな生体分子の機能をできるだけ,それが機能・作用する環境で解析したい,そのためには,細胞小器官(オルガネラ)や細胞骨格が作る「細胞」という器と,その細胞が持っている「細胞内環境」を組み入れた細胞アッセイ系を構築したい.「セミインタクト細胞リシール技術」はそのようなタンパク質や生体分子の機能解析という基礎生命科学と創薬・医療開発における細胞アッセイの要請のもとに生まれた.
2. 生命現象を再構成するためのセミインタクト細胞系
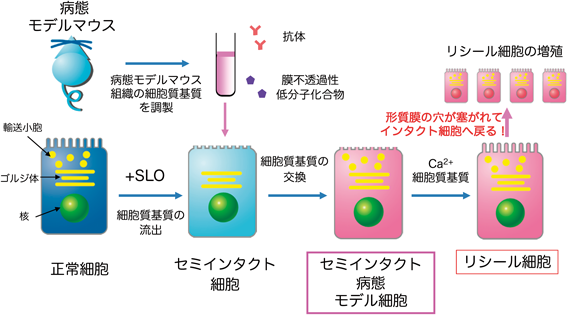
「セミインタクト細胞」とは,レンサ球菌の酸素感受性毒素ストレプトリジンO(SLO)を用いて細胞膜に一時的に孔を開け,細胞質を流出させた細胞のことである1).SLOは,低温(一般的には,4°C)では細胞膜にあるコレステロールに結合するだけであるが,25°C以上に昇温すると,細胞膜上で環状に自己集合することによりチャネル様構造を作る2).つまり,4°CでSLOをコレステロールに結合させ,その後,未結合のSLOを4°Cの緩衝液で洗い流し,続けて細胞系を昇温するという操作により,細胞膜のみに孔を開け,細胞質中にあるオルガネラや細胞骨格の構造はほぼインタクトな状態で維持しながら細胞質基質(サイトゾル)を流出させることができる.このような状態の細胞を「セミインタクト細胞」と呼ぶ(図1).
セミインタクト細胞では,逆に,細胞外から添加したさまざまな分子をこの孔を通過させて細胞内に導入することができる.導入できる物質の種類や大きさは多様であり,タンパク質に限らず,核酸,人工合成・修飾された生体分子,膜不透過性化合物など多岐にわたるさまざまな種類の分子を同時に,しかもその比率をコントロールしながら導入できる.さらに興味深いことに,導入できる分子の数は原理的には制限がないことから,多種類の成分からなる生体分子の混合物である「細胞質基質」をそのまま導入することもできる.たとえば,セミインタクト正常細胞に病態細胞・組織より別途調製した「病態細胞質基質」を導入することにより,セミインタクト細胞内を「病態環境」に改変することも可能である.また,細胞内のオルガネラや細胞骨格の構造や相対的配置はほぼ完全に保持されていることから,セミインタクト細胞内の生体分子[タンパク質,脂質,遺伝子(mRNAなども)]や細胞質基質と一緒に導入されたさまざまな分子プローブの反応をまさにそれが機能する「現場」と「環境」において,導入した細胞質基質依存的に分析的に再構成し,生命現象を駆動・制御する細胞質基質・細胞因子を解析・同定することができる.つまり,セミインタクト細胞とは,細胞膜・オルガネラ・細胞骨格が作る構造的環境(細胞の構造体のintegrityとそれらの相対位置関係)を保持したままで構成要素的環境(細胞質基質)を交換・操作できる「細胞型試験管」である.
我々は,これまでにこのセミインタクト細胞技術とGFP可視化技術を組み合わせたさまざまな細胞アッセイを構築して,光学顕微鏡下の「単一細胞内」で生起する細胞周期依存的な小胞体,ゴルジ体などのオルガネラ形態変化や両オルガネラ間の小胞輸送過程を再構成し,関わる制御因子の同定とその機能解析に利用してきた1, 3–6).
3. 「セミインタクト細胞系」を用いた細胞分裂期(M期)超早期の細胞内イベントの再構成
このセミインタクト細胞を使ったアッセイの特長の一つとして,「生命反応の同期」がある.特定の状態の細胞から細胞質基質を調製し,セミインタクト細胞内に導入することで,1個~106個オーダーの細胞内で生起する反応を「同期して開始させる」ことができるため,一般に五月雨式に進行する細胞内の生化学反応を,良好なS/N比で解析できる可能性がある.ここでは,その代表的研究例として,M期初期に動物細胞内で起こるゴルジ体の形態変化の例を示す.
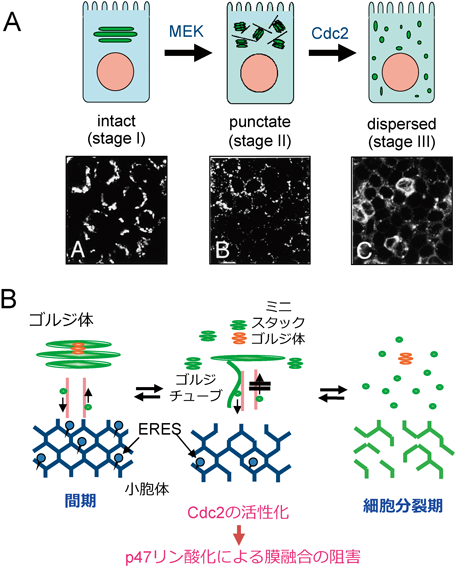
哺乳動物細胞の間期ゴルジ体は,核近傍の層板構造とネットワーク構造からなる特徴的な形態を示す一方,細胞分裂期(M期)には,一度その安定な形態を壊し,娘細胞内に均等分配されそこで再構築される7).一般に,M期に突入した細胞内では,関連キナーゼの活性化・不活化を介して細胞骨格系やオルガネラ形態の変化が協奏的に進行するため,微小管の状態や他のオルガネラ(特に,小胞体など)との小胞輸送に形態形成・維持機構を依存しているゴルジ体の分解(disassembly)過程の解析を生細胞系で行うのは困難をきわめていた.そこで,まず,ゴルジ体に局在する酵素ガラクトシルトランスフェラーゼ(GT)のGFP融合タンパク質(GT-GFP)を恒常的に発現するイヌ腎臓尿細管上皮由来細胞(MDCK-GT細胞)を樹立した.共焦点レーザー顕微鏡によるタイムラプス観察により,インタクトなMDCK-GT細胞では,間期には核の一極にあるリボン状の層板構造を持つゴルジ体(stage I)が,G2期の終わり(M期の直前)ごろにはいったんフラグメント化し約0.8 µmのミニスタックゴルジ体(小さいながらも三層構造を持ったゴルジ膜)を含む少し大きめのゴルジ膜(stage II)へと分解される.さらに細胞分裂期が進むと,フラグメント化したゴルジ体はさらに細かくなり(小胞化し),細胞質全体に分散するようすが観察された(stage III).つまり,M期ゴルジ体の分解過程は,光学顕微鏡レベルでも形態的に三つのstageと二つの素過程に分割できることがわかった(図2A).セミインタクト細胞系は,このように形態的に分割できる複数の素過程それぞれを駆動する細胞質基質因子の解析・同定に威力を発揮する.今回のセミインタクト細胞アッセイ系では,MDCK-GT細胞をセミインタクト細胞にし,M期に同期させたアフリカツメガエル(Xenopus)卵抽出液を「M期細胞質基質」として導入し,M期ゴルジ体の分解過程を再構成した3).同調したM期細胞質基質の大量入手が可能であったカエル卵抽出液を哺乳動物培養細胞内に導入するという荒っぽいやり方であったが,M期細胞質基質を導入したセミインタクトMDCK-GT細胞では,そのゴルジ体が上記の2段階の形態変化を経て小胞化することが確認できた.このことから生化学的な反応を保持する細胞質基質はセミインタクト細胞内で十分にその機能を発揮することがわかった.そこで次に各素過程に必要な細胞質基質因子[ここでは,当時話題になっていた二つのキナーゼ:MEK(MAP kinase kinase 1)8)とcdc2キナーゼ9)]の作用点を生化学的に明らかにすることにした.手法はシンプルである.(i)それぞれのキナーゼ(その機能)を除去した細胞質基質では,ゴルジ体分解は阻害されるか,(ii)間期の細胞質基質でそれぞれのキナーゼを活性化すると,ゴルジ体分解が誘起されるか,を調べることにした.まず(i)については,抗cdc2キナーゼあるいは抗MEK抗体を用いて,M期細胞質基質からそれぞれのキナーゼを免疫除去した細胞質基質を用意し,そのゴルジ体分解に対する効果を調べた.cdc2キナーゼを除去したM期細胞質基質ではstage IIで,MEKを除去したM期細胞質基質ではstage Iでゴルジ体分解が停止した.(ii)については,間期細胞質基質にcdc2キナーゼを活性化させるサイクリンAタンパク質,あるいはMEKを活性化させるSTE1タンパク質を添加することで,それぞれを活性化させた間期細胞質基質を調製し,その効果を調べた.予想どおり,cdc2キナーゼを活性化させた間期細胞質基質ではstage Iで停止し,MEKを活性化させた間期細胞質基質ではstage IIまでゴルジ体分解が進んだ.また先にMEKを活性化した間期細胞質基質を加え,その洗浄後にcdc2キナーゼを活性化した間期細胞質基質を添加した場合,stage IIIまでゴルジ体分解が進んだのに対し,逆の順番で加えた場合は,stage IIで停止した.以上のことから,stage I→IIの素過程にはMEK, stage II→IIIの素過程にはcdc2キナーゼが順番に作用することが示唆された3)(図2A).このように,セミインタクト細胞法では,導入する細胞質基質に対して免疫除去,ドミナントアクティブ・ネガティブ作用を持つリコンビナントタンパク質などを添加するなどして,細胞質基質のもつ機能を修飾することが可能であり,それら生化学的手法を駆使して細胞質基質因子の同定・検証が可能になる.
M期におけるゴルジ体分解過程は,それまで,細胞から単離したゴルジ体を用いin vitro系で再構成されていた.さらに,ゴルジ体の層板間のスタッキングに関わるGRASP6510),GRASP5511)の発見やその制御機構は多くがこのin vitro再構成系から得られた知見である.しかし,ゴルジ体のようにその形態が微小管等の細胞骨格系に強く依存するオルガネラの細胞周期依存的な形態変化の一部は,細胞骨格系をインタクトに近い状態で保持したセミインタクト細胞系を用いることで初めて再現される可能性も考えられる.実際,stage IIで同定されたミニスタックゴルジ体は,MDCK細胞のアピカル側の微小管ネットワークに強く結合した状態で存在しており,生細胞では検出が困難であったMEKの関与を,セミインタクト細胞アッセイ系により浮かび上がらせたことになった.
4. 細胞周期依存的な小胞体ネットワークの形態変化および小胞体−ゴルジ体間小胞輸送の再構成
ゴルジ体と小胞体は小胞輸送によって結ばれており,両オルガネラの形態は小胞輸送のバランスやキネティクス変化に大きく影響される.そこで,両オルガネラの形態が大きく変化するM期の細胞質基質存在下のセミインタクト細胞における小胞体の形態およびゴルジ体–小胞体間小胞輸送について検証を行った.マウス胸腺リンパ腫由来のL5178Y細胞から別途調製したM期細胞質基質をセミインタクト細胞に導入したところ,M期における小胞体ネットワークの部分的切断が形態的に再構成され,さらにこの過程がcdc2キナーゼと,その基質の一つであるp47(膜融合因子p97のコファクター)のリン酸化に依存的であることがわかった5).さらに,主にゴルジ体に局在しているものの,実際は小胞体とゴルジ体間を小胞輸送経路に乗って循環することが知られていたGT-GFPタンパク質を輸送マーカーとし,FRAP(fluorescence recovery after photobleaching)法によって小胞体→ゴルジ体の順行輸送,ゴルジ体→小胞体の逆行輸送を定量したところ,M期細胞質基質存在下では順行輸送は停止するが,逆行輸送は正常かむしろ少し活性化気味に継続することがわかった1, 4).このことは,小胞体ネットワークからゴルジ体に向かう輸送小胞の出芽に関わる「小胞体搬出部位(ER exit site:ERES)」がcdc2キナーゼ依存的にM期細胞質基質によって分解されることと一致する結果であった4).
M期細胞質基質を用いたゴルジ体・小胞体ネットワーク・ERES・両オルガネラ間の小胞輸送の再構成の結果を総合的に判断すると,M期におけるゴルジ体の形態変化は,「ゴルジ体構造制御装置」と「メンブレントラフィック制御装置」の絶妙なカップリングによって起こっていることがわかる(図2B).M期初期に活性化するcdc2キナーゼが,まず,ERESのp47のリン酸化依存的な分解を通して,小胞体からゴルジ体への順行小胞輸送を停止させるが,ゴルジ体からのCOPI依存的な小胞形成と逆行輸送は継続すると思われる.このとき,GRASPsのリン酸化によるゴルジ体層板の「unlinking」も並行して進行する.細胞分裂後の娘細胞では,cdc2キナーゼの不活化とともにこの逆の過程が進行してERES再構築,順行輸送の再開,各オルガネラの再構築が進行すると予想される.
5. セミインタクト細胞を用いたタンパク質のオルガネラ局在化機構の研究
前述したように,細胞内のタンパク質は特定のオルガネラや細胞骨格を最適な機能発現の「場」として局在化している.その仕組みは多様で,核,ミトコンドリア,小胞体に局在化するタンパク質は局在化に必要なタグとなる「シグナル配列」を持つ場合と持たない場合がある.後者の例として,さまざまなオルガネラに局在化しオルガネラ間の小胞輸送やコミュニケーションに関わる単量体GTPase Rabファミリータンパク質がある.Rabファミリータンパク質はヒトで約60種類ものタンパク質群からなる大きなファミリーであり,これらは共通のGTP結合部位と脂質修飾部位を持つが,それぞれのRabタンパク質は異なった細胞内局在を示す12).たとえば,Rab1は小胞体とゴルジ体,Rab2はシスゴルジ体,Rab5はエンドソームに局在し,各オルガネラにおける小胞輸送過程制御に関与する.アミノ酸配列上にはオルガネラ局在化タグ配列はなく,そのオルガネラ局在化制御因子を体系立てて解析する手法は開発されていなかった.我々はセミインタクト細胞系を開発し始めた初期の段階から,このRabタンパク質の局在化制御因子の探索系の開発ができないものかと常に考えていたが,最初のターゲットを動物細胞のゴルジ体に局在化するRab6Aにした.
まず,グルタチオンS-トランスフェラーゼ標識したRab6Aタンパク質(以降GST-Rab6A)を大腸菌で作らせ精製した.セミインタクトHeLa細胞に,精製したGST-Rab6Aタンパク質を加えると,これだけではゴルジ体への局在化は観察できなかった.そこでL5178Y細胞から調製した細胞質基質(+ATP再生系)共存下でGST-Rab6Aを加えると,GST-Rab6Aがゴルジ体へと局在化することが蛍光抗体法により確認できた13).このことはL5178Y細胞質基質中に,GST-Rab6Aをゴルジ体へと局在化させる必要因子があることを強く示唆した.そこで,次にL5178Y細胞質基質からGST-Rab6Aに結合するタンパク質群を免疫共沈降法で網羅的に回収し,質量分析法により同定した.その結果,タンパク質BICD2が同定された.BICD2は微小管モータータンパク質ダイニンと輸送小胞をつなぐアダプタータンパク質の一種であり,実際,BICD2を免疫除去した細胞質基質やBICD2に対する抗体を加えた細胞質基質存在下では,GST-Rab6Aのゴルジ体局在化が阻害されたことなどから,BICD2がRab6Aのゴルジ体ターゲティングに関与することが確認された.また,興味深いことに,微小管のintegrityはこのBICD2依存的なGST-Rab6Aのゴルジ体ターゲティングには影響がなかった.つまり,BICD2は微小管モータータンパク質のアダプターとは別の機序でこのターゲティングに関わっている可能性も示唆された13).
ここで確立したRab6Aタンパク質のゴルジ体局在化因子探索法は,他のRabタンパク質ファミリーの細胞内局在化制御因子を探索することにも簡単に応用できるとともに,Rabタンパク質ファミリー以外の細胞質タンパク質のオルガネラ局在化機構研究にも汎用的に応用できる.この研究は,「細胞型試験管」のセミインタクト細胞アッセイ系の特長がうまく活かせた好例となっている.
細胞内構造を保持し,細胞内環境を改変できるセミインタクト細胞系は,細胞の形態・場所情報とそれと関連する分子情報を元に生命現象を再構成し分析するときに威力を発揮する.しかし,セミインタクト細胞系には,その構築原理に根ざした大きな問題もある.それは,細胞膜に孔が空いたままでの解析システムであることである.細胞膜上に形成されている孔のため細胞内外の区別が厳密でないことにより,細胞膜を介したエンド・エキソサイトーシスの膜動過程,細胞膜上の受容体を介したさまざまな増殖・分化シグナルの細胞質から核内への伝達過程などの解析系としては問題が多い.
このようなセミインタクト細胞アッセイの問題点を意識し始めたころ,作製したセミインタクト細胞の細胞膜が修復され(孔が再封入され),実験中に生細胞に戻ることを我々はたびたび経験していた.実際,SLOによって形成された細胞膜上の孔は,カルシウムイオン依存的に誘起されたエクトサイトーシスやエンドサイトーシスなどの膜動過程により除去・修復されるという報告もある14–16).そこで,我々は,セミインタクト細胞に別途調製した細胞質基質を導入し,細胞膜にあいた孔をカルシウム依存的に再度閉じる「リシール細胞」技術の開発とリシール細胞の標準化を目指した(図1,リシール細胞).
リシール細胞技術に眼が向いたのは,当時の生命科学の大発見が発端となっている.それは山中伸弥博士のiPS細胞の発見である.我々は,タンパク質の寿命制御機構を組み込んだ山中4因子をリコンビナントタンパク質として調製し,リシール細胞技術を利用して細胞内導入することで,ウイルスベクターを使用しない安全なiPS細胞を創成できると考えていた.同時に,未分化性を保持した細胞質基質の導入や,iPS細胞の分化誘導に最適な細胞質基質の導入も考えていた.同様な試みは世界的に始まっていた.同じころ,ノルウェーのCollasらのグループは,我々と同様なリシール細胞技術を用いて,293T細胞に胚性腫瘍細胞抽出液を導入し,Oct4やNanogのDNAメチル化やヒストンの修飾などのエピジェネティックな変化を誘導したり17),また,T細胞抽出液を導入し293T細胞をT細胞に分化誘導させたりする実験を報告した18).リシール細胞技術を用いたリコンビナントタンパク質やES細胞抽出液の導入によるiPS細胞作製も当然ながら報告され始めていた19, 20).山中4因子のリコンビナントタンパク質を調製しMEF細胞にリシール細胞技術で導入すると,数日後には確かにいくつかのiPS細胞マーカーの発現が上昇し,エピジェネティックな変化も観察されたが,その万能性を維持できず1週間も経つとほとんどが脂肪や神経細胞に分化してしまった.4因子の発現のタイミング調節法を吟味しないままに始めたプロジェクトの苦い結果であった.
残念な結果となったリシール細胞技術のiPS細胞作製への応用であったが,いくつかの重要な知見も同時に得られた.(i)リシール細胞に導入された細胞質基質の性質は,導入された細胞内で約12時間以上保持されること,(ii)導入した細胞質基質によって細胞内の核が一時的に曝露されることで,エピジェネティクス動態や遺伝子発現変動が誘起されること,(iii)一度のリシール操作で約106個のリシール細胞を調製し高効率で生存・増殖させられること,などである.特に,(iii)については,多くの研究グループはiPS細胞作製時には,iPS細胞の絶大な増殖能を利用し,偶発的に生じた少数の(極端には単一細胞でも)細胞を特定の条件でスクリーニングすることでiPS細胞を樹立する手法をとっていたが,我々は,愚直に,できるだけ均一で大量のリシール細胞を得ることに専心し,かつその生存率を上げるためのシステムを追求し続けていた.このことは,このリシール細胞系を生化学的解析や多くの網羅的解析(オミックス解析)に供することができることも意味していた.
我々は,ヒト子宮頸がん由来培養細胞(HeLa細胞)をプロトタイプとしてリシール細胞作製技術の標準化を進めた21).リシール細胞作製効率や導入物質の細胞内保持率は,蛍光標識デキストランの細胞内への導入効率とその細胞内保持能を指標にして,蛍光顕微鏡あるいはフローサイトメトリーを用いて定量的に比較検討できる.その定量化手法を用い,リシール化の条件(CaCl2濃度,必要細胞質基質濃度,作用時間など)について詳細に検討した.また,リシール細胞の生存率は,セミインタクト細胞作製条件に大きく依存することから,セミインタクト細胞化の条件(SLO処理時の濃度,時間,細胞密度など)も検討した.これら検討項目を標準化し,HeLa細胞以外のさまざまな細胞系に対してセミインタクト細胞リシール技術の標準化を実施できるようになり,今では,リシール細胞条件を数日で決定できるようになっている.ちなみに,HeLa細胞のリシール操作は,1 mM CaCl2で3分以上のインキュベーションで十分誘導でき,細胞質基質は>1.5 mg/mLの濃度でリシールを促進することが明らかになった.この最適化された条件では,HeLa細胞において90%以上という高いシール効率と生存率を達成できた.最適な条件によって作製されたリシール細胞は,形態・機能的に生きているHeLa細胞とほとんど大差ない状態を維持し,増殖能も変化なかった.つまり,「セミインタクト細胞リシール技術」を駆使することにより,細胞内に導入されたタンパク質(群)のさまざまな動的平衡状態を「真の細胞環境内に近い状態で」計測できる可能性が高くなった.
8. 「細胞編集ツール」としてのセミインタクト細胞リシール技術
セミインタクト細胞リシール技術の利用法は,まさしく「細胞内物質導入(デリバリー)法」であるが(たとえば,in cell NMRへの応用22, 23)),同時に,導入物質による「細胞内や核内ニッチ(環境)の改変・同調法」であり,「改変細胞質環境におけるタンパク質や生体分子の作用機序解析」までを含んでいる.我々は,このリシール細胞作製技術を利用し,さまざまな細胞を「編集し,評価してみたい」と考え始めた.そしてその最初の試みは,「病態モデル細胞」にすることを研究開始の初期段階で決めていた.
なぜ「病態モデル細胞」が必要なのか? 正常および病態の網羅的マイクロアレイ解析やDNAメチル化解析,プロテオーム解析が進み,多数の疾患関連遺伝子(産物)が抽出されている.しかし,抽出される病態関連遺伝子(産物)の数は膨大であり,動物個体を使用した創薬・診断の前臨床研究のコストと時間が大幅に増加している.そのため,「細胞」を用いた新規の病態誘発遺伝子・タンパク質の絞り込み法の開発はますます重要となってきている.
最も現実に即した環境で細胞レベルの薬効を調べるためには,患者から採取した病態を反映する細胞を用いるのがベストではあるが,現実には初代培養細胞の寿命に限りがある点や不均一性(正常細胞やさまざまな病態進行過程の細胞が混在している)があるという点で患者由来の細胞は扱いにくく,このような細胞集団を用いた疾患マーカーの同定や,薬効の評価は現実的には困難である.一方,実験室で一般的に用いられる培養細胞の多くはがん化した細胞であり,目的にかなった特定の疾患表現型を示す培養細胞は稀有である.また,近年では患者由来のiPS細胞を基に作製した疾患モデル細胞を用いた個別化医療・創薬が期待されているが,iPS細胞が得意とするのは主に遺伝性疾患であり,生活習慣などが長い期間をかけて影響するような疾病の研究には必ずしも適当ではない.そこで,我々の研究グループでは生活習慣病の一つである糖尿病に着目し,セミインタクト細胞リシール技術を用いた病態モデル細胞の作製と疾患マーカー同定,そして糖尿病改善化合物のスクリーニングを行うことにした.
糖尿病は日本国内で300万人以上が罹患する生活習慣病である.糖尿病ではインスリンの作用が減弱しているため,慢性的に血糖値が高まる.膵β細胞から分泌されるホルモンであるインスリンは,細胞に作用するとグルコースの取り込みを促進するが,糖尿病患者では膵β細胞からのインスリン分泌が少なくなるのみならず,肝臓・脂肪・筋肉などでインスリンが存在するにもかかわらずグルコースが取り込めない「インスリン抵抗性」を獲得してしまう.このように,細胞レベルのインスリン抵抗性という病態がそのまま個体全体や組織の病態となって現れるため,我々の研究室では膵臓,脂肪,肝臓において糖尿病で不全となるさまざまな生命現象について細胞レベルでの研究を行ってきた24–29).これらの研究を通して蓄積されたノウハウや知見に加え,2型糖尿病はゲノム変異のみではなく生活習慣による長期間の細胞ストレスの影響が蓄積する結果であるという点からも,リシール細胞技術を利用した病態モデル細胞として最適な研究対象であると考えた.
9. リシール細胞の作製から初期の「病態モデル細胞」作製へ
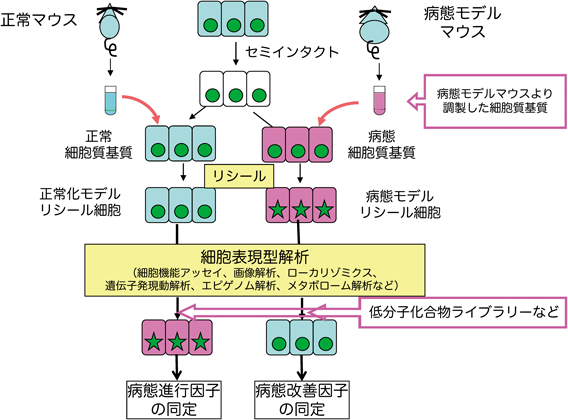
まずは,糖尿病モデル動物(db/dbマウス)の肝臓などの組織から調製された「糖尿病態細胞質基質」をセミインタクトHeLa細胞内に導入して「細胞内を糖尿病態環境に同期」した「糖尿病モデル細胞」を作製した21).同時に,コントロールとして正常モデルマウスから同じ手法で調製した「正常細胞質基質」を用いて,「正常モデル細胞」も作製した.ここで作製された「正常モデル細胞」と「糖尿病モデル細胞」は,同じ操作でリシール操作を経てHeLa細胞株に導入されたため,この2種類のモデル細胞の性状の「差分」を詳細に解析することで,リシール細胞作製のための操作によるアーティファクトは相殺され,逆に糖尿病態細胞質基質由来の特異的な細胞現象や細胞内の分子ネットワークの違いが抽出されてくるはずであると考えた.このように,正常と病態モデル細胞の2種類を作製し,その差分をさまざまな解析(ローカリゾミクス,遺伝子発現変動解析,エピゲノム解析,メタボローム解析など)結果から抽出することで,S/N比の良好な糖尿病特有の細胞の表現型を精度よく検出することができた.そして,この2種類の細胞質基質の違いを持ったリシール細胞の「差分」から注目する生命現象に関わる分子ネットワークを抽出する手法が,今では,リシール細胞解析の基本戦略となっている(図3).
まず,正常および糖尿病モデル細胞について,さまざまなオルガネラの形態変化やそこに局在化するタンパク質の違いを蛍光抗体法を用い網羅的に解析したところ,糖尿病モデル細胞では,初期エンドソームに局在する脂質の一つであるホスファチジルイノシトール三リン酸(PI3P)が,正常モデル細胞に比べて極端に減少していることを発見した21).初期エンドソームにPI3Pが少ない原因を探っていくと,糖尿病モデル細胞内では酸化ストレス状態になっているため,ストレス応答キナーゼp38 MAPKの活性化が起こり,その結果,PI3Pを作る酵素Vps34がエンドソーム膜にリクルートされないことが原因の一つであることがわかった30).初期エンドソームの細胞質側膜表面に局在するPI3Pは,細胞質からさまざまなタンパク質をリクルートして,初期エンドソームでの物質選別(ソーティング)制御に関わっており,この制御機構の撹乱は糖尿病態発現のさまざまな原因になることが十分予想される.たとえば,PI3P量の減少は,初期エンドソームにおけるSmadアンカータンパク質(SARA)のエンドソームへのリクルートを妨げることで,TGFβ/Smad系のシグナル伝達に影響を及ぼす31).また,糖尿病モデル細胞では,成長因子(EGF)添加前からEGF受容体だけがすでに細胞内に選択的に取り込まれてエンドソームやリソソームまで移行し分解されていることが確認された.これは糖尿病モデル細胞の細胞膜上のEGF受容体の数がもともと減少しており,結果的に細胞が受けうる成長シグナルが受けにくいことを示唆し,実際,糖尿病マウスの肝臓においても,EGF受容体数の減少が検出されており32),糖尿病発現との関係が精力的に研究されている.我々の作製した糖尿病モデル細胞を利用すれば,その初期エンドソームの機能的撹乱と細胞膜上のEGF受容体減少の因果関係を通し,糖尿病態発現の分子ネットワークを細胞レベルで解明できると期待して研究を進めている.
10. 糖尿病モデル肝細胞の表現型解析と創薬スクリーニング
これまで主にHeLa細胞などの汎用化される細胞でリシール細胞系を構築してきたが,肝臓由来の正常・糖尿病細胞質基質を肝臓由来の細胞に導入することで,より生体に近い糖尿病の肝臓細胞状態が再現されると予想される.この糖尿病モデル肝臓細胞で糖尿病の典型的な表現型である糖代謝異常などが再現できれば,糖尿病を標的にした創薬や治療のためのより有用な細胞側ツールとなりうる.そこで肝臓から調製した細胞質基質を導入する細胞として,インスリンに応答して糖新生酵素の発現抑制がみられるラット肝がん由来培養細胞H4IIEC3細胞を用いることにした.H4IIEC3細胞をセミインタクト細胞にし,正常あるいは糖尿病モデルマウス肝臓の細胞質基質を導入後,リシールした.それぞれの正常あるいは糖尿病モデル肝細胞における糖代謝について,i)糖新生酵素PCK1とG6PCの発現,ii)培地中グルコース量を定量し検証した.すると正常モデル細胞ではインスリン依存的な糖新生酵素の発現抑制がみられたのに対し,糖尿病モデル肝細胞では発現抑制が阻害されていることが明らかになった33).また培地中グルコース量についても,正常モデル肝細胞ではインスリン依存的に減少しているのに対し,糖尿病モデル肝細胞では増えて異常な状態となっていた.これらの結果から,まさに糖尿病モデル肝細胞を用いて,実際の肝臓組織で起こっている糖尿病態である「インスリン抵抗性」(インスリン依存的な糖新生抑制の撹乱)が再現できたと考えている.
構築した正常・糖尿病モデル肝細胞の性状を詳細に調べたところ,インスリンシグナル伝達において中心的な役割を果たすAktの活性化(リン酸化)が阻害されていることを見いだした33).このAktリン酸化阻害はインスリン応答異常の原因となりうるのみならず,糖尿病モデル肝細胞特異的な表現型,すなわち「疾患マーカー」として活用することができる.
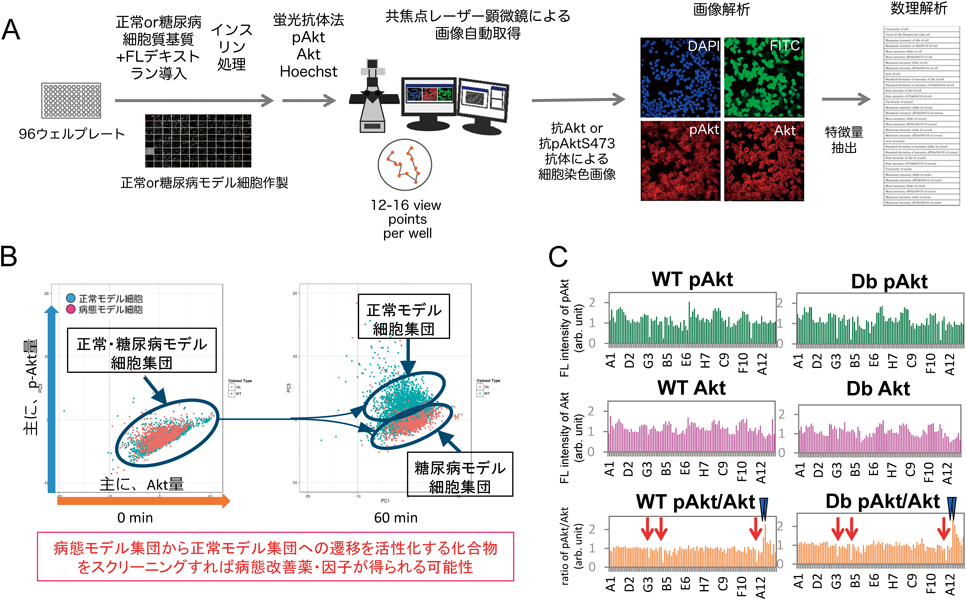
細胞内環境を同期した大量の病態モデル細胞を作製することにより,多様な網羅的解析や生化学的な解析が可能になる.しかし,大量の細胞を処理するときにはどうしてもリシール操作による細胞質基質交換効率が不均一になる可能性が生じる.また,リシールの標的となる細胞の種類によっては,リシール効率が非常に悪い細胞種がある可能性は否めない.そこで,我々は,リシール細胞の表現型解析に,光学顕微鏡下に観察できる単一細胞の画像解析をベースにしたアプローチを実施した.ウエスタンブロッティング法などの生化学的方法では細胞集団全体の平均的な量しか判別できないのに対し,画像解析法では,抗Akt抗体と抗リン酸化Akt(S473)抗体を用いた蛍光抗体法により,蛍光顕微鏡下に観察される単一の細胞1個1個におけるAkt量とリン酸化Akt(S473)量の比をある程度定量的に求めることが可能である.具体的には,96ウェルプレートに正常あるいは糖尿病モデル細胞を作製し,インスリン処理を行った.細胞を固定し,抗Akt抗体と抗リン酸化Akt(S473)抗体を用いた間接蛍光抗体法を行い,細胞内のAktおよびpAktS473を蛍光でラベルした.共焦点レーザー顕微鏡を用いて自動撮影により数十万個レベルの細胞画像を取得し,単一細胞ごとの細胞全体,核,細胞質領域におけるAktあるいはpAktの蛍光強度(平均,分散,最大,最小など)や核の円形度などの構造についての数値を「特徴量」として抽出した(図4A).これらの特徴量を用いて主成分分析を行ったところ,正常・糖尿病モデル肝細胞を分けうる指標として「pAkt/Akt比」が適していることを見いだした(図4B).さらにpAkt/Akt比を指標に,薬として市販されている化合物90種類のライブラリーと糖尿病治療にも用いられる7種類の化合物について上記の画像ベースのスクリーニングを行った.その結果,正常・糖尿病モデル細胞のpAkt/Akt比を上昇(病態→正常)あるいは低下(正常→病態)させる化合物を同定した(図4C).pAkt/Akt比を減少させる三つの化合物(アバシミブ,クリゾチニブ,PF-431396)はすでに治療薬として使用されている化合物であるため,肝臓のインスリン応答をより糖尿病態へと近づける副作用を持つ可能性が示唆された.またpAkt/Akt比を上げる二つの化合物(メトホルミンとピオグリタゾン)は実際の糖尿病治療薬であり,このような化合物が抽出されてきたことはアッセイの妥当性を示唆すると考えている.また,ピオグリタゾンは糖尿病モデル細胞でよりpAkt/Akt比を上昇させたが,画像解析から得られたデータからpAkt/Akt比が上がるのはリン酸化Akt蛍光量が上昇するからではなく,Akt蛍光量が減少するためであることがわかった.ピオグリタゾンが細胞内のAkt蛍光量を減少することが細胞の表現型にどのような影響を与えるのかは未知であるが,本病態モデル細胞は薬の作用機序を細胞レベルで解明するために有用な細胞側ツールであることを示すことができたと考えている.
これまでみてきたように,細胞質基質はリシール操作やリシール細胞の生存・生育に欠かせないものである.しかし,最近になって,この細胞質基質の必要性を再考せねばならない事態が起こりつつある.
一つ目は,CRISPR/Cas9システムを用いたゲノム編集技術へのリシール細胞技術の応用時に現れた.ゲノム編集(相同組換えやエピジェネティクス改変など)を達成したリシール細胞は,最終的に個体(ヒト,マウス,ブタなど)に再導入してその遺伝子矯正や細胞機能賦活化の効果を検証する必要があるが,そのとき,素性の判然としない「細胞質基質」を持ち込むことへの懸念である.この要請を受けて,我々は,現在,急ピッチでリシール操作やリシール後の細胞の生存に必要な細胞質基質因子の探索を行っている.その結果,予想しなかった複数の細胞質基質因子や低分子化合物がその細胞質基質の代替品となる可能性も出てきた.今後は,これらのリコンビナントタンパク質を用いた最適な「細胞質基質代替品」を使える時代が来るかもしれない.
二つ目は,中分子創薬の隆盛である.特殊ペプチドや核酸などの分子量1000~1万程度の中分子が,新しいバイオ医薬品として注目を集めてきている.その候補中分子医薬品の多くは細胞膜不透過性であり,in vitroでの活性確認はできてもさまざまなタンパク質や生体分子が高濃度に存在する細胞内での効能を確認するのは簡単ではない.中分子の設計段階から膜透過性機能を持たせる設計を行うことや,cell penetrating peptide(CPP)のような膜透過性を持つ短鎖ペプチドタグを目的中分子に付加するなど,さまざまな工夫がなされているが,まずは,in vitroで活性を持つ中分子を細胞内に導入し,細胞内でその効能を検証することで,細胞内デリバリー戦略が立てやすくなる.この細胞内への中分子導入と導入分子の細胞内機能アッセイを,リシール細胞系を使って簡便に行うことが検討されてきた.しかし,SLOを使った通常のリシール技術は,わざわざ大きな孔を細胞膜に開けて細胞質基質を流出させ,中分子と一緒に細胞質基質を戻してリシール操作を行う必要がある.そこで,中分子導入だけが目的の「細胞質基質非依存的なリシール細胞技術」を構築することにした.研究室でさまざまな細胞毒素を試した結果,SLOと同様な性質を持つcholesterol-dependent cytolysin(CDC)の一種であるリステリオリジンO(LLO)を見つけることができた34).この毒素を適当な条件で細胞膜に作用させると,中性条件下で,しかも細胞質基質非依存的に膜不透過性中分子を細胞内に導入し,カルシウム依存的にリシールすることができることがわかった.しかも,SLO型のリシール細胞技術に比較して細胞膜のダメージが圧倒的に少なく,操作も非常に簡単であるため,ハイスループットなスクリーニング系にも十分応用できる可能性もある.実際,このLLO型リシール法を利用して,Aktのキナーゼ活性を阻害することが知られている膜不透過性ペプチドAkt-inの細胞内機能を検証した.その結果,Akt-inにCPPの一種TATペプチドを結合させた膜透過性TAT-Akt-inと同等の,細胞内活性[Aktキナーゼの活性化(リン酸化)阻害]を示せることがわかった34).我々の開発した新規中分子細胞内導入とその機能解析法(LLO型リシール法)は,従来のSLO型リシール法と組み合わせることで,一度,作製したリシール細胞にその後次々とさまざまな中分子を連続的に導入し,新しい細胞の表現型を創出できる技術としても利用できる可能性がある.
「リシール細胞」の特長は,まさに細胞質基質交換によって生細胞になることであり,解析目的の生命現象が再構成しやすいという点にあった.しかし,逆に,リシール操作終了と同時に,細胞内の反応が生細胞様に動き始めるため,多くの生命反応が一気に進むことになり,再構成法の利点である「素過程」の分析的再構成が難しくなることをしばしば経験する.セミインタクト細胞系では,協奏的に進む生命反応を生化学的に,形態的にいくつかの「素過程」に分割し分析することで,当該生命反応の制御因子の同定や検定,反応中間体・構造体などの検出が可能となる.本稿最初に紹介した細胞周期依存的なオルガネラ形態変化やオルガネラ間の小胞輸送の再構成とその素過程の制御機構の解析例は,まさしく,素過程解析に適したセミインタクト細胞系利用の好例である.つまり,セミインタクト細胞系とリシール細胞系(SLO型とLLO型)は,目的に応じて適宜補完的に併用することが必要であることがわかる.
セミインタクト細胞リシール技術の汎用性は高く,細胞の種類や導入する細胞質基質の種類を変えることで,さまざまな病態モデル細胞や分化・発生モデル細胞の構築とそれを利用した制御因子解析に利用できる.そのため,リシール細胞の表現型解析や細胞内の分子ネットワーク解析法開発は,作製したリシール細胞の正確な「設計」と「評価」を行うために必要不可欠な開発項目にもなってきている.たとえば,本稿で紹介したリシール効率のヘテロ性の問題を補償するために開発した「光学顕微鏡を用いた単一細胞の定量的画像解析技術」による病態モデル細胞表現型解析技術は,その一例である.この他にも,我々の研究室では,多くの共同研究を通して,細胞形態の定量的イメージング画像から,細胞内の「分子ネットワーク」を導出する新しい画像解析技術を開発している.その画像解析技術をセミインタクト細胞リシール技術と最適に組み合わせることで,定量的な分子情報に,形態情報をも組み入れた新しい生化学の誕生に貢献できればと思っている.
謝辞Acknowledgments
セミインタクト細胞リシール技術は,「かたち」と「分子」のつながりを見つけたいさまざまな分野の研究者や技術者の方々に支えられてきました.そして,今では,その見つけたつながりを「細胞編集」に応用したい社会に育てられつつあります.この技術の育成と応用に期待しながら関わった・関わる多くの方々に深く感謝致します.
引用文献References
1) Kano, F. & Murata, M. (2013) The semi-intact cell system and methods for cell resealing: A novel systems biology tool to elucidate protein networks with spatio-templioral information. Advances in Systems Biol., 2, 6–14.
2) Bhakdi, S., Tranum-Jensen, J., & Sziegoleit, A. (1985) Mechanism of membrane damage by streptolysin-O. Infect. Immun., 47, 52–60.
3) Kano, F., Takenaka, K., Yamamoto, A., Nagayama, K., Nishida, E., & Murata, M. (2000) MEK and Cdc2 kinase are sequentially required for Golgi disassembly in MDCK cells by the mitotic Xenopus extracts. J. Cell Biol., 149, 357–368.
4) Kano, F., Tanaka, A.R., Yamauchi, S., Kondo, H., & Murata, M. (2004) Cdc2 kinase-dependent disassembly of endoplasmic reticulum (ER) exit sites inhibits ER-to-Golgi vesicular transport during mitosis. Mol. Biol. Cell, 15, 4289–4298.
5) Kano, F., Kondo, H., Yamamoto, A., Tanaka, A.R., Hosokawa, N., Nagata, K., & Murata, M. (2005) The maintenance of the endoplasmic reticulum network is regulated by p47, a cofactor of p97, through phosphorylation by cdc2 kinase. Genes Cells, 10, 333–344.
6) Kano, F., Kondo, H., Yamamoto, A., Kaneko, Y., Uchiyama, K., Hosokawa, N., Nagata, K., & Murata, M. (2005) NSF/SNAPs and p97/p47/VCIP135 are sequentially required for cell cycle-dependent reformation of the ER network. Genes Cells, 10, 989–999.
7) Valente, C. & Colanzi, A. (2015) Mechanisms and regulation of the mitotic inheritance of the Golgi complex. Front. Cell Dev. Biol., 3, 79.
8) Acharya, U., Mallabiabarrena, A., Acharya, J.K., & Malhotra, V. (1998) Signaling via mitogen-activated protein kinase kinase (MEK1) is required for Golgi fragmentation during mitosis. Cell, 92, 183–192.
9) Lowe, M., Rabouille, C., Nakamura, N., Watson, R., Jackman, M., Jämsä, E., Rahman, D., Pappin, D.J., & Warren, G. (1998) Cdc2 kinase directly phosphorylates the cis-Golgi matrix protein GM130 and is required for Golgi fragmentation in mitosis. Cell, 94, 783–793.
10) Barr, F.A., Puype, M., Vandekerckhove, J., & Warren, G. (1997) GRASP65, a protein involved in the stacking of Golgi cisternae. Cell, 91, 253–262.
11) Shorter, J., Watson, R., Giannakou, M.E., Clarke, M., Warren, G., & Barr, F.A. (1999) GRASP55, a second mammalian GRASP protein involved in the stacking of Golgi cisternae in a cell-free system. EMBO J., 18, 4949–4960.
12) Stenmark, H. (2009) Rab GTPases as coordinators of vesicle traffic. Nat. Rev. Mol. Cell Biol., 10, 513–525.
13) Matsuto, M., Kano, F., & Murata, M. (2015) Biochim. Biophys. Acta–Mol. Cell Res., 1853, 2592–2609.
14) Idone, V., Tam, C., Goss, J.W., Toomre, D., Pypaert, M., & Andrews, N.W. (2008) Repair of injured plasma membrane by rapid Ca2+-dependent endocytosis. J. Cell Biol., 180, 905–914.
15) Keyel, P.A., Loultcheva, L., Roth, R., Salter, R.D., Watkins, S.C., Yokoyama, W.M., & Heuser, J.E. (2011) Streptolysin O clearance through sequestration into blebs that bud passively from the plasma membrane. J. Cell Sci., 124, 2414–2423.
16) Atanassoff, A.P., Wolfmeier, H., Schoenauer, R., Hostettler, A., Ring, A., Draeger, A., & Babiychuk, E.B. (2014) Microvesicle shedding and lysosomal repair fulfill divergent cellular needs during the repair of streptolysin O-induced plasmalemmal damage. PLoS One, 9, e89743.
17) Freberg, C.T., Dahl, J.A., Timoskainen, S., & Collas, P. (2007) Epigenetic reprogramming of OCT4 and NANOG regulatory regions by embryonal carcinoma cell extract. Mol. Biol. Cell, 18, 1543–1553.
18) Håkelien, A.M., Landsverk, H.B., Robl, J.M., Skålhegg, B.S., & Collas, P. (2002) Reprogramming fibroblasts to express T-cell functions using cell extracts. Nat. Biotechnol., 20, 460–466.
19) Zhou, H., Wu, S., Joo, J.Y., Zhu, S., Han, D.W., Lin, T., Trauger, S., Bien, G., Yao, S., Zhu, Y., et al. (2009) Generation of induced pluripotent stem cells using recombinant proteins. Cell Stem Cell, 4, 381–384.
20) Bru, T., Clarke, C., McGrew, M.J., Sang, H.M., Wilmut, I., & Blow, J.J. (2008) Rapid induction of pluripotency genes after exposure of human somatic cells to mouse ES cell extracts. Exp. Cell Res., 314, 2634–2642.
21) Kano, F., Nakatsu, D., Noguchi, Y., Yamamoto, A., & Murata, M. (2012) A resealed-cell system for analyzing pathogenic intracellular events: perturbation of endocytic pathways under diabetic conditions. PLoS One, 7, e44127.
22) Yamaoki, Y., Kiyoishi, A., Miyake, M., Kano, F., Murata, M., Nagata, T., & Katahira, M. (2018) The first successful observation of in-cell NMR signals of DNA and RNA in living human cells. Phys. Chem. Chem. Phys., 20, 2982–2985.
23) Ogino, S., Kubo, S., Umemoto, R., Huang, S., Nishida, N., & Shimada, I. (2009) Observation of NMR signals from proteins introduced into living mammalian cells by reversible membrane permeabilization using a pore-forming toxin, streptolysin O. J. Am. Chem. Soc., 131, 10834–10835.
24) Fujiki, K., Kano, F., Shiota, K., & Murata, M. (2009) Expression of the peroxisome proliferator activated receptor gamma gene is repressed by DNA methylation in visceral adipose tissue of mouse models of diabetes. BMC Biol., 7, 38.
25) Fujiki, K., Shinoda, A., Kano, F., Sato, R., Shirahige, K., & Murata, M. (2013) PPARγ-induced PARylation promotes local DNA demethylation by production of 5-hydroxymethylcytosine. Nat. Commun., 4, 2262.
26) Sugawara, T., Kano, F., & Murata, M. (2014) Rab2A is a pivotal switch protein that promotes either secretion or ER-associated degradation of (pro)insulin in insulin-secreting cells. Sci. Rep., 4, 6952.
27) Arai, T., Kano, F., & Murata, M. (2015) Translocation of forkhead box O1 to the nuclear periphery induces histone modifications that regulate transcriptional repression of PCK1 in HepG2 cells. Genes Cells, 20, 340–357.
28) Nakatsu, D., Horiuchi, Y., Kano, F., Noguchi, Y., Sugawara, T., Takamoto, I., Kubota, N., Kadowaki, T., & Murata, M. (2015) L-cysteine reversibly inhibits glucose-induced biphasic insulin secretion and ATP production by inactivating PKM2. Proc. Natl. Acad. Sci. USA, 112, E1067–E1076.
29) Horiuchi, Y., Nakatsu, D., Kano, F., & Murata, M. (2017) Pyruvate kinase M1 interacts with A-Raf and inhibits endoplasmic reticulum stress-induced apoptosis by activating MEK1/ERK pathway in mouse insulinoma cells. Cell. Signal., 38, 212–222.
30) Kano, F., Arai, T., Matsuto, M., Hayashi, H., Sato, M., & Murata, M. (2011) Hydrogen peroxide depletes phosphatidylinositol-3-phosphate from endosomes in a p38 MAPK-dependent manner and perturbs endocytosis. Biochim. Biophys. Acta, 1813, 784–801.
31) Miura, S., Takeshita, T., Asao, H., Kimura, Y., Murata, K., Sasaki, Y., Hanai, J.I., Beppu, H., Tsukazaki, T., Wrana, J.L., et al. (2000) Hgs (Hrs), a FYVE domain protein, is involved in Smad signaling through cooperation with SARA. Mol. Cell. Biol., 20, 9346–9355.
32) de Diego, J.G., Rouiller, D.G., Gorden, P., & Carpentier, J.L. (1992) Epidermal growth factor receptor internalization and biosynthesis in the diabetic rat. Exp. Cell Res., 200, 77–82.
33) Kano, F., Noguchi, Y., & Murata, M. (2017) Establishment and phenotyping of disease model cells created by cell-resealing technique. Sci. Rep., 7, 15167.
34) Murakami, M., Kano, F., & Murata, M. (2018) LLO-mediated Cell Resealing System for Analyzing Intracellular Activity of Membrane-impermeable Biopharmaceuticals of Mid-sized Molecular Weight. Sci. Rep., 8, 1946.
著者紹介Author Profile
 村田 昌之(むらた まさゆき)
村田 昌之(むらた まさゆき)東京大学大学院総合文化研究科教授.東京大学・社会連携講座「次世代イメージング画像解析学講座」特任教授.東京工業大学科学技術創成研究院細胞制御工学研究センター特任教授.理学博士.
略歴1988年京都大学大学院理学研究科博士課程修了.89年京都大学大学院理学研究科助手(この間ドイツ・ヨーロッパ分子生物学研究所,米国・UCバークレーにて客員研究員),96年より岡崎・生理学研究所助教授,2003年より現職.
研究テーマと抱負次世代イメージング画像解析技術とセミインタクト細胞リシール法を主軸にした「細胞設計と編集」,そしてその細胞の「評価」を高速で循環させ,細胞を理解し利用する「細胞デザイン」拠点創成を目指しています.
ウェブサイトhttp://muratalab.c.u-tokyo.ac.jp/index.html
趣味カフェ巡り.
 加納 ふみ(かのう ふみ)
加納 ふみ(かのう ふみ)東京工業大学科学技術創成研究院准教授.理学博士.
略歴2002年京都大学大学院理学研究科博士課程修了.03年より東京大学大学院総合文化研究科助教.その間07~11年,11~15年にJSTさきがけ研究員兼任.16年より現職.
研究テーマと抱負セミインタクト細胞リシール法の開発とそれを用いた細胞質交換による疾患モデル細胞の構築,創薬スクリーニングなどの応用研究.細胞の形質・機能を制御しうるテクノロジーとしての「細胞編集工学」を確立します.
ウェブサイトhttp://kanolab.rcb.iir.titech.ac.jp
趣味ピアノを再開したいです.