自閉症の分子メカニズムMolecular mechanism in autism spectrum disorder
理化学研究所脳神経科学研究センターRIKEN Center for Brain Science ◇ 〒351–0198 埼玉県和光市広沢2–1 ◇ 2–1 Hirosawa, Wako, Saitama 351–0198, Japan
発行日:2018年8月25日Published: August 25, 2018
自閉症は近年増加の一途をたどり,社会的な問題となっている.自閉症は遺伝率が高いことから発症の原因としてゲノム変異が示唆されているが,必ずしも遺伝的に説明がつくわけではない.さらに自閉症の候補遺伝子は数百に上り,その機能的な多様性からも分子メカニズムを解明することはいまだに困難である.本稿では,ゲノムワイドな遺伝学研究から検出されてきた自閉症のゲノム変異に基づいて,遺伝子機能の共通点から自閉症の分子メカニズムを議論する.
© 2018 公益社団法人日本生化学会© 2018 The Japanese Biochemical Society
自閉症は神経発達障害の一つであり,現在は自閉スペクトラム症(autism spectrum disorder:ASD,以下ASDと表記する)と呼ばれる.ASDの主な症状として,社会的行動や対人コミュニケーションに支障を来すことや,繰り返し行動や特定のものごとに強いこだわりを示すことがあげられる.また,光や音の刺激に対して過敏もしくは鈍感,他人からの物理的な接触を嫌うなど,感覚系の異常を有することが特徴である.さらに,それぞれの症状の程度は個々人によって幅広い差があり,スペクトラム(英語spectrum:範囲や連続体など幅を持った分布を意味する)として捉えるようになった.ASDは米国精神医学会による精神疾患の診断・統計マニュアル第5版(DSM-5)をもとに,医師によってその症状が診断される.DSM-IVまで広汎性発達障害のサブカテゴリーとして定義されていた自閉症やアスペルガー症候群などの分類がDSM-5で廃止され,レット症候群を除くすべての障害名がASDに統一された.1943年にLeo Kanner博士によって最初の臨床報告がなされたASDは,近年,日本のみならず世界的に患者数が増加しており,1990年代では1000人に2人ほどであった発症率が,2014年の米国の例では59人に1人といった高い発症率が報告されている.しかしながら,その原因は明らかとなっておらず,発症率増加は社会的な問題としても議論されている.行動学的にはスペクトラムとして捉えられるように,診断基準の拡張によってASDと診断される人が増えた可能性が考えられる.また,遺伝学的には出産年齢の高齢化に伴う弧発性変異の増加が影響している可能性なども考えられるが,その直接的な証拠はない.1970年代を始めとして30年以上にわたる双子の臨床研究から,自閉症では遺伝的な要因が高いことが示唆されている.Bourgeronの総説によると,一卵性双生児の共発症率が36~95%に及ぶのに対し,兄弟間や二卵性双生児の共発症率は0~38%程度である1).共通したゲノム情報を有する一卵性双生児の高い共発症率は,ASDの発症には遺伝的要因が大きく関与していることを示唆する.遺伝学的研究からはASDの遺伝的素因の多様性が見いだされている.近年では,この遺伝的な多様性がスペクトラムとしての行動表現型の多様性につながり,さらには効果的な治療方法の発見を妨げている原因ではないかと考えられている.また,環境因子の影響も否定されていない.妊娠期のウイルス感染などによる免疫機能の過活動,また,抗てんかん薬や抗うつ薬の使用は,胎児の脳発達また将来のASD症状に影響すると考えられている.本稿では,ASDの分子メカニズムを捉えるため,ゲノム解析研究から見つかったASDにおける遺伝的変異の共通性とそれら変異を再現したヒト型モデルマウス研究を取り上げて解説する.
遺伝学的研究からは,ASD患者に存在する多数のゲノム変異が報告されている2).これらは親から子へ遺伝するタイプの遺伝性変異と,親には存在せず子だけが持つタイプのde novo変異(弧発性変異,精子や卵子の配偶子形成時に偶発的に起こるもの)に分類される.古典的には細胞遺伝学的手法によって染色体を可視化し,核型を比較することで,ゲノム変異が発見されてきた.この手法では実体顕微鏡下で確認できるほどの比較的大きな染色体異常(7q21-22, 7q31-32, 15q11-13, 22q11.2などの重複/欠失や逆位重複)が検出される3).近年の技術の発展に伴い,アレイCGH(比較ゲノムハイブリダイゼーション)と呼ばれる全ゲノムのコピー数の増幅や欠損を包括的に調べる技術や,ゲノムワイド関連解析(GWAS)と呼ばれるゲノム全体にわたる50~100万か所の一塩基多型を解析する技術によって小さな染色体異常も検出できるようになった.さらに次世代シーケンサーの発展に伴い,mRNAに転写されたエクソン領域を読み取る全エクソーム配列解析(whole exome sequencing)やゲノム情報すべてを読み取る全ゲノム配列解析(whole genome sequencing)によって,ゲノム上のわずか数塩基の違いも検出することができ,個人のゲノムを網羅的に解析することが可能となった(表1).ゲノム変異のうち,コピー数多型(copy number variation:CNV)はゲノム上の数kbから数MbのDNA領域が欠失または重複する変異である.CNVの及ぼす影響として,CNVが遺伝子領域全体を含有することで,その領域に含まれる遺伝子発現量がコピー数依存的に増減されることやエクソンの一部を含有することで遺伝子機能が消失されることが生じる.米国シモン財団のASDゲノム変異のデータベースSFARIに登録されているCNVの数は2000以上に上る.このうち健常者にも共通して見つかっているCNVも存在するため,現在130~250ほどのCNV領域がASD関連領域として示唆されている4, 5).また,一塩基多型(single nucleotide variation:SNV)は,遺伝子の塩基配列内に変異が生じるもので,コーディング領域ではタンパク質のアミノ酸配列を置換させる(ミスセンス変異)または終止コドン化させる(ナンセンス変異)ことでタンパク質の機能を失わせる.コーディング領域外では,プロモーターやエンハンサー領域といった遺伝子発現機構を変化させることが影響として現れる.また数塩基~数十塩基の挿入や欠損によってフレームシフトが生じる変異もASDの全エクソーム配列解析で発見されている.先述のSFARIには990遺伝子が登録されており,このうち複数のゲノム解析研究から得られたデータをもとに統計学的に信頼性の高いものとして,カテゴリー1“High Confidence”に24遺伝子,カテゴリー2“Strong Candidate”に59遺伝子がASD関連遺伝子として示されている(表2).このようにASDを対象として確認されているゲノム変異は千のオーダーで存在するが,この中からASD発症に結びつく遺伝子を同定することはなかなか困難である.その理由は遺伝的変異の多様性であり,また,健常者の中にも同様の変異を持つ人が存在することである.遺伝的変異の影響は,その含まれる遺伝子の数や種類によってさまざまであり,ASD症状の重症度は影響の大きい遺伝的変異によるか影響の小さい遺伝的変異によるかに依存する(図1A).主要なCNVの例をあげると,15q11-13重複はASD群で348例見つかっているがコントロール群では見つかっていない.一方,16p11.2欠失/重複はASD群で1159例,コントロール群で316例見つかっている6).また,複数の報告例があるものでも,中には4q32.1欠失/重複のようにASD群134例,コントロール群120例といった割合で見つかるCNVもある.このように特定の遺伝的変異はASD発症と必ず相関することもあれば部分的に相関することもある.ASD発症に対して特に影響力の強い変異はレアバリアントと呼ばれ,健常者にも見つかるような影響力の弱い変異はコモンバリアントと呼ばれている.これまでのゲノム研究から,レアバリアントを主とした遺伝的変異がASDの直接的原因であると見積もられている割合は,ASD全体を100%としてCNVが5~10%,SNVが4~9%である(図1B).残念ながらCNVとSNVは合わせても全体の2割ほどしかなく非常に少ない.ASD患者の8割程度は遺伝学的な原因探索が達成できていない.そのためASDの研究者たちは,複数のコモンバリアントもしくは環境要因が組み合わされた結果,複合的な要因の積み重ねがASD発症の原因の大多数を占めるのではないかと推定している.それでは,どのようにすれば複雑なASDの分子メカニズムを探索できるのか? まずはASD発症と相関するような影響力の強いレアバリアント遺伝子を調べ,そこから派生する遺伝子ネットワークの機能的変動を探ることが基準となる.
| 文献 | 解析対象 | サンプル | 数 | 主な発見 |
|---|---|---|---|---|
| Sebatら2007 | CNV | ゲノムDNA(血液,B細胞) | 264家系(118単発性家系,47多発性家系,99コントロール家系),195ASD患者,196健常者 | ASD関連CNV:14 |
| Pintoら2010 | CNV | ゲノムDNA(血液,細胞系列) | 996ASD患者,1287健常者 | ASD関連CNV:226 |
| ASD関与遺伝子:36 | ||||
| ASD関与遺伝子座:10 | ||||
| Sandersら2011 | CNV | ゲノムDNA(血液,唾液,細胞系列) | 1174家系(計4457人) | ASD関連CNV:130–234 |
| Pintoら2014 | CNV | ゲノムDNA | 2446家系(Pinto 2010のデータを含む) | 病因性CNV:84 |
| Iossifovら2012 | SNV | ゲノムDNA(血液) | 343家系 | ASD:59 SNV |
| コントロール:28 SNV | ||||
| FMRP関連遺伝子 | ||||
| O’Roakら2012 | SNV | ゲノムDNA(血液) | 209家系(計677人) | ASD関連遺伝子:62 |
| βカテニンおよびクロマチン制御遺伝子 | ||||
| Sandersら2012 | SNV | ゲノムDNA(血液,細胞系列) | 238家系(計928人) | de novo SNV:279 |
| Iossifovら2014 | SNV | ゲノムDNA(血液) | 2517家系(2508 ASD患者,1911健常者) | クロマチン制御遺伝子,FMRP関連遺伝子 |
| Voineaguら2011 | 遺伝子発現 | 死後脳組織 | 前頭葉:16ASD患者,16健常者 | シナプス機能遺伝子,免疫反応遺伝子 |
| 側頭葉:13ASD患者,13健常者 | ||||
| 小脳:11ASD患者,10健常者 | ||||
| Guptaら2014 | 遺伝子発現 | 死後脳組織 | 47ASD患者,57健常者 | 活性型ミクログリア遺伝子,シナプス伝達遺伝子 |
| Ginsbergら2012 | 遺伝子発現とメチル化DNA | 死後脳組織 | BA19:9ASD患者,9健常者 | ASD特異的メチル化領域の非検出 |
| 発現減少:ミトコンドリア酸化的リン酸化遺伝子,タンパク質翻訳 | ||||
| Ladd-Acostaら2014 | メチル化DNA | 死後脳組織 | 19ASD患者,21健常者 | 4遺伝子座のASD特異的メチル化領域 |
| Nardoneら2014 | メチル化DNA | 死後脳組織 | BA10:12ASD患者,12健常者 | 低メチル化:免疫反応遺伝子 |
| BA24:13 ASD患者,2健常者 | 高メチル化:シナプス遺伝子 | |||
| Nardoneら2017 | メチル化DNA | 死後脳組織 | BA10:15ASD患者,16健常者 | シナプス伝達,シナプス可塑性,神経投射,GABA代謝経路,GABAシグナル,免疫反応 |
| CNV: copy number variation, SNV: single nucleotide variation, BA: Brodmann area | ||||
| シンボル | 遺伝子名 | 染色体領域 | カテゴリー | タンパク質機能 | レアバリアント/コモンバリアント |
|---|---|---|---|---|---|
| ADNP | Activity-dependent neuroprotector homeobox | 20q13.13 | 1, S | クロマチンリモデリングによる遺伝子発現制御 | 43/0 |
| ANK2 | Ankyrin 2, neuronal | 4q25-q26 | 1 | 細胞骨格,細胞移動,細胞増殖 | 69/0 |
| ARID1B | AT rich interactive domain 1B (SWI1-like) | 6q25.3 | 1, S | ATP依存的クロマチンリモデリング複合体の構成分子 | 92/0 |
| ASH1L | Ash1 (absent, small, or homeotic) -like (Drosophila) | 1q22 | 1 | トリソラックス群転写活性化複合体の構成分子 | 37/0 |
| ヒストンメチル基転移酵素 | |||||
| ASXL3 | Additional sex combs like 3 (Drosophila) | 18q12.1 | 1, S | ポリコーム群転写抑制化複合体の構成分子,ホメオティック遺伝子の転写抑制 | 52/0 |
| CHD8 | chromodomain helicase DNA binding protein 8 | 14q11.2 | 1 | DNAヘリカーゼ,βカテニンと相互作用して転写抑制 | 77/0 |
| クロマチンリモデリング | |||||
| CUL3 | Cullin 3 | 2q36.2 | 1 | cullinタンパク質ファミリー,ポリユビキチン化,タンパク質分解 | 16/0 |
| DSCAM | Down syndrome cell adhesion molecule | 21q22.2 | 1 | 免疫グロブリンスーパーファミリーに属する細胞接着分子 | 15/1 |
| DYRK1A | Dual-specificity tyrosine-(Y)-phosphorylation regulated kinase 1A | 21q22.13 | 1, S | 細胞増殖シグナル経路の制御因子 | 103/0 |
| GRIN2B | glutamate receptor, inotropic, N-methyl D-apartate 2B | 12p13.1 | 1, S | NMDA受容体サブタイプ | 123/32 |
| KATNAL2 | Katanin p60 subunit A-like 2 | 18q21.1 | 1 | 微小管供給,微小管の再組織化 | 16/0 |
| KMT2A | Lysine (K) -specific methyltransferase 2A | 11q23.3 | 1, S | 転写活性化因子 | 33/0 |
| メチル基転移活性 | |||||
| KMT5B | lysine methyltransferase 5B | 11q13.2 | 1 | ヒストンメチル基転移酵素 | 14/0 |
| H4K20メチル化,転写抑制 | |||||
| MYT1L | Myelin transcription factor 1-like | 2p25.3 | 1, S | 神経転写因子 | 34/1 |
| 神経分化 | |||||
| 神経およびオリゴデンドロサイト | |||||
| NAA15 | N (alpha) -acetyltransferase 15, NatA auxiliary subunit | 4q31.1 | 1 | アセチル基転移酵素 | 22/0 |
| 血管,造血,神経成長 | |||||
| POGZ | Pogo transposable element with ZNF domain | 1q21.3 | 1, S | ジンクフィンガータンパク質 | 73/0 |
| 細胞分裂促進,動原体形成,姉妹染色分体の接着 | |||||
| PTEN | phosphatase and tensin homolog | 10q23.31 | 1, S | がん抑制 | 66/0 |
| ホスファチジルイノシトール-3,4,5-三リン酸3-ホスファターゼ | |||||
| RELN | Reelin | 7q22.1 | 1, S | 分泌型細胞外マトリックスタンパク質,細胞–細胞間相互作用,神経細胞移動 | 91/7 |
| SCN2A | sodium channel, voltage-gated, type II, alpha subunit | 2q24.3 | 1, S | 電位依存性イオンチャネル | 193/0 |
| SETD5 | SET domain containing 5 | 3p25.3 | 1, S | メチル基転移酵素 | 68/0 |
| SHANK3 | SH3 and multiple ankyrin repeat domains 3 | 22q13.33 | 1, S | 足場タンパク質 | 233/8 |
| SYNGAP1 | synaptic Ras GTPase activating protein 1 | 6p21.32 | 1, S | PSD構成分子NMDA受容体関連 | 84/0 |
| TBR1 | T-box, brain, 1 | 2q24.2 | 1 | 転写制御分子 | 19/1 |
| TRIP12 | Thyroid hormone receptor interactor 12 | 2q36.3 | 1 | PA700構成分子 | 35/0 |
| 26S複合体のATP依存的タンパク質分解活性 | |||||
| S:症候性カテゴリー(特定の症候性障害に含まれる遺伝子) | |||||
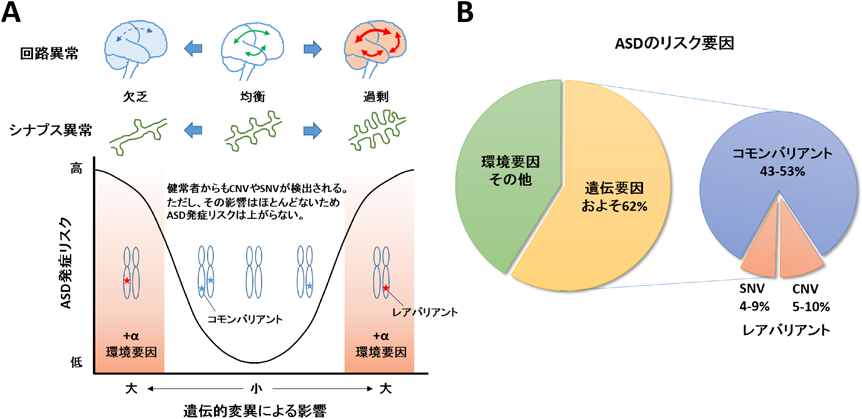
(A) ASD患者からは健常者に比べて約3倍多くゲノム変異が見つかっており,タンパク質機能を失わせるような影響力の大きい変異を持つことが多い.ASD発症に対して影響力の強い変異は検出される確率が低いためレアバリアントと呼ばれており,逆に健常者にも見つかるような影響力の弱い変異は頻繁に検出されるためコモンバリアントと呼ばれる.ASDで見つかるゲノム変異は多様であり,ASD患者それぞれ異なるため遺伝的な原因を特定することは難しい.中には健常者と同じコモンバリアントしか持たないASD患者も存在する.また,遺伝要因のみならず環境要因によってASD発症リスクが増加することが示唆されている.表現型としてのASD症状はスペクトラムであり,境目が明瞭ではない.遺伝要因や環境要因の多様性が問題であり,各個人がどのようなリスク因子を有するのかを知ることが,ASD発症メカニズムを理解するために必要である.また,ASDの表現型の一つとしてシナプス異常が注目されている.シナプスが過剰形成または欠乏することで神経回路としての機能障害を生じる.過不足のどちらの方向であっても,均衡状態から外れることがASD発症につながると考えられている.(B) ASDにおけるリスク要因の割合.CNVやSNVなどのゲノム変異が原因でASD発症につながる割合は全体の約2割である.その他は複数の遺伝要因,環境要因が相乗的に作用することでASD発症につながると考えられている.
遺伝的素因に多様性が存在するとしても,結果的にASD発症につながるのであれば,脳内で起こる分子メカニズムには共通点が存在すると多くの研究者たちは考えている.ASD患者のゲノム変異が及ぼす分子ネットワークとしての機能障害を探ることで,ネットワークレベルで対処するような効果的な治療方法を見いだせるかもしれない.
1990年代以前から近年に至るまでに,死後脳の解剖学的研究によってASD患者の神経細胞の樹状突起スパイン(主に興奮性入力を受けている樹状突起上の棘状構造)の形態異常が報告されてきた7).そのためASD病態にスパインやシナプス(神経細胞どうしの接続.神経細胞が発火したときに生じる活動電位の電気シグナルを神経伝達物質の化学シグナルに変換して,次の神経細胞に情報伝達する構造)の異常が関係しているのではないかと示唆されてきた8).さらに2000年代前半にはASD患者を持つ家系の遺伝学的解析により,ASD患者ではシナプス構造に関わるタンパク質をコードする遺伝子(NLGN3, NLGN4X, SHANK3)にゲノム変異があることが報告された9, 10).さらにはNLGN3変異やSHANK3欠失を再現したモデルマウス研究において,これらのモデルマウスがシナプスの機能異常を有することや行動学的にもASD様症状を示すことが明らかとなった11, 12).そのため,ASDにおけるシナプス遺伝子異常がASD発症における重要なキーワードとなるのだが,遺伝学の発展は単純な答えを導き出したわけではなかった.2000年代後半から2010年代にかけて,技術発展による大規模なゲノム解析が行われることとなり,数多くのCNVやSNVが新たに見つかった.そして,これらのCNVやSNVに含まれる遺伝子群の特徴が明らかとなってきた(図2).
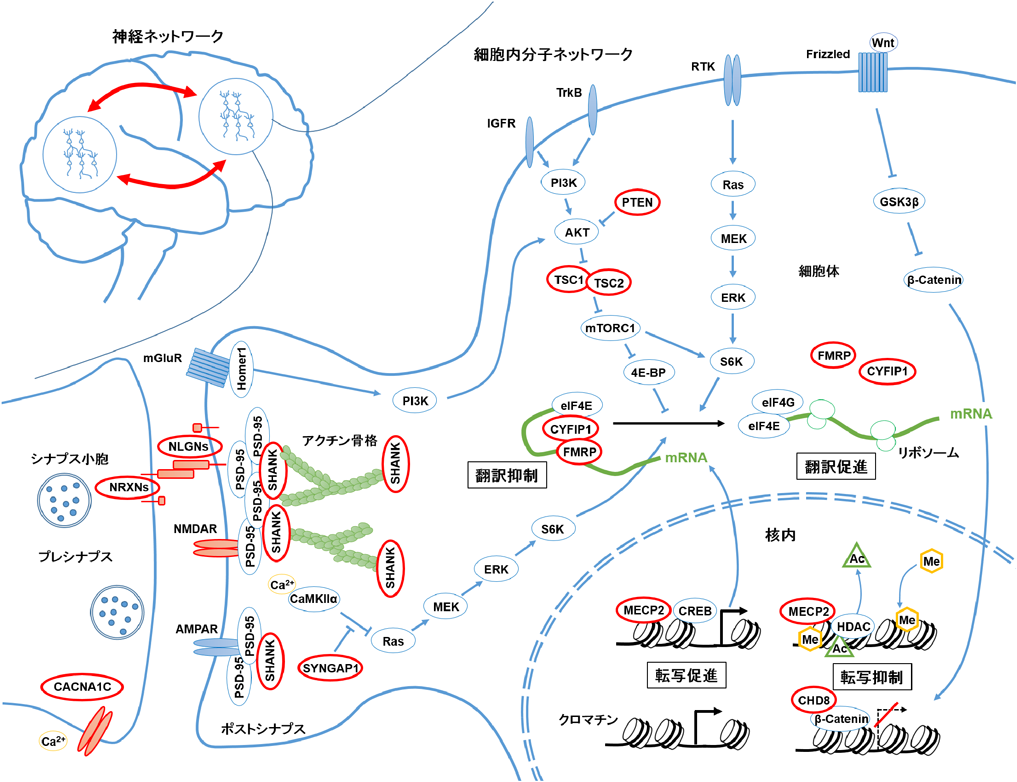
ASDの神経病態学的メカニズムには,ミクロレベルからマクロレベルまで幅広いネットワーク機能障害が関与している.ゲノム研究から,シナプスや遺伝子発現機構に関わる分子ネットワークの異常が示唆されている.ASD患者ではクロマチン制御遺伝子のゲノム変異が頻出している.CHD8やMECP2は,主に核内でヘテロクロマチン形成に関わり,標的遺伝子の転写を抑制する.これらの遺伝子にゲノム変異が生じると転写抑制機能が失われるため,予期しない遺伝子発現が起こる.また,翻訳制御に関わる遺伝子のゲノム変異も頻出しており,代表的なPTEN, TSC1/2, FMRPは翻訳を抑制する働きを持つ.そのため,ゲノム変異でタンパク質機能が失われると,翻訳機構が活性化され,過剰にタンパク質合成が起こる.これらの遺伝子発現機構が破綻すると,神経細胞の正常発達が阻害され,神経ネットワークの機能性が低下する.ASD脳では,特にシナプス分子の遺伝子発現変動が顕著である.転写翻訳抑制に関わる遺伝子のゲノム変異では,シナプス分子の過剰合成がスパイン過剰形成につながっていると考えられる.また,シナプス構成分子(NRXN, NLGN, SHANK)のゲノム変異も多数見つかっており,モデルマウス研究においてこれらの機能障害はシナプス結合やシナプス機能を低下させることが確認されている.図1で示したように,シナプスが過剰形成されても欠乏しても神経ネットワーク機能障害を生じる.細胞内分子ネットワークを正常化することで領域間の神経ネットワークを改善させることができると期待される.ASD関連遺伝子を赤丸(太枠)で示した.AKT:protein kinase B (PKB, 別名AKT), CACNA1C:calcium channel, voltage-dependent, L type, alpha 1C subunit, CaMKIIa:Ca2+/calmodulin-dependent protein kinase II alpha, CHD8:chromodomain-helicase-DNA-binding protein 8, CREB:cAMP response element binding protein, CYFIP1:cytoplasmic FMRP-interacting protein 1, eIF4E:eukaryotic translation initiation 4E, ERK:extracellular signal-regulated kinase, FMRP:fragile X mental retardation protein, GSK3β:glycogen synthase kinase 3 beta, IGFR:insulin-like growth factor receptor, MECP2:methyl-CpG binding protein 2, MEK:MAPK/ERK kinase, mGluR:metabotropic glutamate receptor, NLGN:neuroligin, NRXN:neurexin, PI3K:phosphoinositide 3-kinase, PI3K:phosphoinositide 3-kinase, PSD-95:postsynaptic density protein 95, PTEN:phosphatase and tensin homologue, RTK:receptor tyrosine kinase, S6K:S6 kinase, SHANK:SH3 and multiple ankyrin repeat domains protein, SYNGAP1:synaptic RAS GTPase-activating protein 1, TrkB:tropomyosin-related kinase B, TSC1/2:tuberous sclerosis 1/2. AC:アセチル基,Me:メチル基.
表1に記載したようにゲノム研究から,130~250ほどのCNV領域がASD関連領域として示唆される.CNVは大きいものでは数Mbにも及ぶので,一つのCNV領域には複数の遺伝子が含まれることが多い.2446家系のゲノム変異を調べたPintoらの研究では,ASD群,コントロール群合わせて6859のCNVが検出された13).de novo CNVの検出率は,ASD群では4.7%,コントロール群では1~2%である.そのうちASD群で検出された102のde novo CNVに含まれる遺伝子について,バイオインフォマティクスを用いたネットワーク解析を行うと,シナプス形成およびシナプス機能,クロマチンリモデリングおよび転写制御に関連する遺伝子群が有意に検出された.一方,コントロール群のde novo CNV解析では有意な遺伝子群は検出されなかった.このことから,ASD発症に関わるCNVにはシナプスや転写制御の機能に影響を及ぼす遺伝子が多く含まれていることが明らかとなった.
主要なCNVモデルとして,15q11-13重複や16p11.2欠失/重複のマウスモデルが染色体工学的手法を用いて作製された14, 15).15q11-13重複領域には,シナプス機能に関わる遺伝子として,GABA受容体遺伝子GABRA5, GABRB3, GABRG3,ユビキチンE3リガーゼ遺伝子UBE3Aなどが含まれる.15q11-13重複マウスは社会性行動などのASD様行動異常のみならず,発達期の脳内におけるセロトニン系異常が報告されている15, 16).神経伝達物質の一つであるセロトニンは神経発達や情動行動に関わることが知られており,セロトニン異常を発達期に是正することによって,15q11-13重複マウスの社会性行動を含む表現型の改善がみられた(5節参照)17).さらに15q11-13重複マウスでは,抑制シナプス入力の減少やスパインのターンオーバー頻度の増加が報告されており,神経ネットワークの機能異常が示唆されている17, 18).16p11.2領域には,転写因子MAZ, TBX6,クロマチン制御因子HIRIP3, INO80E,タンパク質キナーゼMAPK3,リボ核タンパク質因子MVPなどが含まれている.16p11.2欠失/重複マウスでは顕著な社会性行動異常は確認されていないが,16p11.2欠失マウスでは感覚系の応答異常や鳴声頻度の減少が報告されている14, 19).16p11.2欠失/重複患者のリンパ芽球細胞系とモデルマウスの大脳皮質を用いた遺伝子発現解析では,シナプス関連NRXN1, NRXN3,クロマチンリモデリング関連CHD8, EHMT1, MECP2,転写制御関連TCF4, SATB2,翻訳制御関連FMR1など多くのASD候補遺伝子が16p11.2のCNVで発現変動を示すことが報告された20).
次にIossifovらのSNVの研究を紹介する.こちらも2500家系を超えるゲノム変異を全エクソン配列解析で調べた結果,ASD群,コントロール群合わせて5648個のde novo SNVが検出され,353個の遺伝子変異箇所がASD発症に影響を与える候補として示された21).これらのSNVが含まれる遺伝子群には,クロマチン制御関連遺伝子,FMRPターゲット遺伝子が有意に検出された.この研究では検出されたSNVにレアバリアントとして知られるいくつかのシナプス関連遺伝子(ANK2, GRIN2Bなど)が含まれるが,SNV遺伝子の機能的分類解析ではシナプス関連遺伝子群に有意差は検出されなかった.また,データベースから複数の文献で検出されたSNV情報を集めて,バイオインフォマティクス的手法で解析を行った研究報告を紹介する22).LiuらはASD患者に特異的に高頻度で検出されるSNV遺伝子に加え,それらと時空間共通性の高い発現パターンを示す遺伝子を集めてネットワーク解析を行ったところ,127個のASD関連遺伝子候補が同定され,それらのタンパク質相互作用に基づく遺伝子オントロジー解析では,七つに分類されるクラスター[Cluster(C)1:転写制御,C2:転写制御およびクロマチンリモデリング,C3:細胞接着および細胞移動,C4:転写制御およびRNAポリメラーゼII,C5:リガーゼ活性,C6:足場タンパク質および受容体シグナリング,C7:翻訳制御]が検出された.このうちC6にはシナプス構造やシナプス伝達に関わる遺伝子が含まれている.
以上のようにASDのゲノム解析では,神経機能やシナプス分子に直接関与する遺伝子のみならず,遺伝子発現に影響するようなクロマチンリモデリングや転写制御機構の遺伝子にゲノム変異が多く検出された.また,特定のCNVによってはそのCNV領域に含まれないASD関連遺伝子発現が影響されることも示されており,ゲノム変異によって引き起こされる遺伝子ネットワーク変動を網羅的に調べることが重要である.
ゲノム変異が脳機能に与える影響を直に調べることは難しい.シナプス関連遺伝子に変異が生じた結果,シナプス形成に異常が生じることや神経細胞機能に障害が生じることは想定できる.しかし,クロマチンリモデリングや転写制御に関わる遺伝子に変異が生じた場合,どのような機能障害が生じるのであろうか? それは脳全体に影響が出るのだろうか? それとも脳領域や細胞の種類によって異なるのであろうか? クロマチンリモデリングや転写制御に関わる遺伝子は他の多くの遺伝子発現に影響を与える.成長過程における遺伝子発現パターンが正常な範囲から逸脱することで,ASDの脳機能発達が妨げられていると予想される.ここではASD患者の死後脳組織から遺伝子発現解析を行った研究を紹介する.
Voineaguらは,ASD群とコントロール群から58皮質検体,21小脳検体を用いてマイクロアレイによる遺伝子発現を解析した23).その結果,皮質では444遺伝子,小脳では2遺伝子がASD検体で有意に変動していた.また,これらの遺伝子と共通した発現パターンを示す遺伝子グループを検索するバイオインフォマティクスのネットワーク解析では,二つのグループがASDに関わる遺伝子群として検出された.一つはM12グループ(シナプス機能,小胞輸送,神経投射),もう一つはM16グループ(免疫・炎症反応,アストロサイト,活性化ミクログリア)である.M12グループはASD脳で発現が減少しており,逆にM16グループは増加している.また,M12グループにおける遺伝子ネットワークのハブ遺伝子とみなされるA2BP1は神経特異的スプライシング制御因子であり,その標的遺伝子にはASD関連遺伝子として報告されているATP5C1, ATP2B1, GRIN1, MEF2Cが含まれる.さらにGuptaらは,ASD群47検体とコントロール群57検体のサンプルから遺伝子発現解析を行い,活性化ミクログリアと免疫反応遺伝子がASDで特徴的な発現変動遺伝子であることを示した24).このようにASD患者の脳内遺伝子発現の特徴として,シナプス関連遺伝子の発現減少と免疫反応やミクログリア遺伝子の発現上昇が見いだされてきた.
また,遺伝子発現に間接的に関わるDNAメチル化状態を調べた研究報告においても,同様の傾向がみられる.Nardoneらの2014年の報告では,ASD群,コントロール群それぞれ12皮質検体[ブロードマン領野(BA)10とBA24]からDNAメチル化状態を解析したところ,免疫関連遺伝子の低メチル化,シナプス遺伝子の高メチル化が確認された25).また,Nardoneらの後続研究としてASD15検体,コントロール16検体のBA10の神経細胞のDNAメチル化状態を解析したエピジェネティクス研究では,シナプス,免疫関連の遺伝子に加えて,抑制伝達物質GABA関連の遺伝子領域にもDNAメチル化状態の変化が捉えられた26).これらは脳内遺伝子発現解析の結果に近く,ASD脳内では免疫関連遺伝子の低メチル化状態が過剰発現を促し,シナプス遺伝子の高メチル化状態が発現抑制を促していることが示唆される.一方,BA19や小脳検体からのDNAメチル化解析では,ASD特異的なメチル化状態の変化は検出されなかった27, 28).これら死後脳検体を用いたDNAメチル化解析の研究報告はサンプルサイズが小さいため統計学的なデータとしての価値は高くない.また,報告によって異なるDNAメチル化状態の変化は,検体として用いた脳領野間の違いに影響されたのかもしれない.
これまでのASD死後脳を用いた遺伝子発現解析は皮質など領野レベルの解析が主体であり,細胞レベルの解像度はなかった.Wangらは,神経細胞,アストロサイト,ミクログリア,オリゴデンドロサイト,神経前駆体細胞などの種類を識別できるシングルセルRNAシーケンサーによる遺伝子発現解析のデータベースをもとに,SFARIなどに登録されている約700のASD関連遺伝子の発現パターンをバイオインフォマティクスで解析した29).その結果,ASD関連遺伝子の多くは神経細胞に豊富に発現しており,特に抑制性神経細胞に多く発現していることが明らかとなった.さらに,抑制性神経細胞に発現しているASD関連遺伝子の多くはコントロールに比べて発現量が増加しており,ASDでは抑制性神経細胞が遺伝子発現変動による影響を受けやすいことが示唆された.また,先行研究と一致して,発現増加を示すASD関連遺伝子の多くは,アストロサイトやミクログリアにも豊富に含まれていた.一方,ASD患者の血液サンプルを用いた解析ではこのような遺伝子発現変動は確認されなかったことから,ASD脳内で生じている神経機能障害は神経細胞特異的な遺伝子発現変動,特に抑制性神経細胞の異常に起因すると考えられる.
遺伝子発現変動やDNAメチル化状態は環境要因によっても影響を受ける.環境要因としてASD発症リスクに影響すると考えられている因子に,妊娠時におけるウイルスまたは病原菌感染,妊娠または出産時の低酸素虚血状態が含まれる30, 31).それらはどのように胎児の脳機能に影響を及ぼすのだろうか.
ウイルス感染などによって母体の免疫反応が過活動になったときの状態をモデル化したマウスの研究では,ヒトの妊娠4~5週目に対応する時期と妊娠28~29週目に対応する時期の妊娠マウスに炎症作用を引き起こすPoly(I:C)を投与し,それぞれの妊娠時期における免疫システムの過活動が胎仔に与える影響を調べた32).産仔が成長してから行動試験を行った後,前頭前皮質のDNAメチル化状態を調査した.ワーキングメモリーを調べるY迷路試験では,後期炎症マウスで成績が減少し,社会性行動は両時期の炎症マウスで低下した.また,感覚情報処理機能の指標とされるプレパルス抑制(先に弱い刺激を加えることで,突然与えられた強い刺激に対する驚愕反応が抑制される現象)は前期炎症マウスで低下した.これらの炎症モデルマウスでは,GABA神経発達とGABAシグナリング(Dlx1, Lhx5, Lhx8など),Wntシグナリング(Wnt3, Wnt8a, Wnt7b),神経発達(Efnb3, Mid1, Nlgn1, Nrxn2など)の各遺伝子群でDNAメチル化状態が変化していた.遺伝子発現変動ではDlx1, Nlgn1は発現減少,Wnt3は発現上昇がみられた.このように妊娠期の炎症によってDNAメチル化状態が変化することで,ASD関連遺伝子の分子ネットワークに含まれる遺伝子発現が影響を受けることが明らかとなった.環境要因によって遺伝子発現変動が起こることで神経発達・神経機能に異常が生じ,結果的に行動異常につながったのではないかと考えられる.
また,ウイルス感染などに対する免疫で分泌されるサイトカインは体内の炎症反応を促進し,脳にも影響する可能性がある.サイトカインの中には,シナプス伝達機能に作用するものが知られている.インターロイキン(IL)-1βは海馬のグルタミン酸放出やNMDA受容体を阻害し,AMPA受容体の膜局在化やリン酸化を阻害するため,LTP(long term potentiation,長期増強:シナプスの伝達効率が強化され,長期的に維持されること)の誘導を妨げる33–35).IL-6はERKシグナル経路を阻害し,LTP誘導を妨げる36).また,腫瘍壊死因子(TNF-α)もp38MAPK経路でLTPを阻害する37).ヒトの死後脳検体や生体から採取した脳脊髄液においてもIL-6やIFN-γを含む多くのサイトカインの上昇が確認されている38).これら炎症性サイトカインがASD脳内でシナプス機能に影響を与えている可能性は十分に考えられる.
原因遺伝子が明らかとなっている精神発達障害患者のうちASDと診断される患者が多いため,それら原因遺伝子もASD関連遺伝子として考えられている.脆弱X症候群のFMR1(5~60%),結節性硬化症のTSC1およびTSC2(約30%),ティモシー症候群のCACNA1C(60~80%),皮質異形成-焦点てんかん症候群のCNTNAP2(約70%)が代表的な例である(パーセンテージはその患者のうちASDと診断される割合).レット症候群はASDと区別されているが,その原因遺伝子MECP2はASD関連遺伝子としてみなされている.
ゲノム変異から遺伝子発現変動に至るまでをまとめると,「クロマチン制御および転写制御に関わる遺伝子群が,発達期におけるシナプス機能および免疫反応に関わる遺伝子発現を乱し,正常な脳神経発達を阻害する」といった過程が想定される.さらに,これらの遺伝子ネットワークに該当する遺伝子であれば,どの遺伝子に変異が生じても,どの遺伝子に発現変動が生じても脳神経発達に及ぼす影響は免れない.ASDを理解するためには遺伝子ネットワークの機能動態を解明することが重要である.
CHD8はCHD(chromodomain, helicase, DNA binding)ファミリーの一つで,ASDのゲノム変異として最も高頻度に検出されている遺伝子である.CHDファミリーはATP依存的にクロマチン構造をリモデリングする.ドメイン構造から三つのサブファミリー(CHD1~2, CHD3~5, CHD6~9)に分類され,どの遺伝子もユビキタスな発現パターンを示すが,CHD3は胎児脳,CHD5は脳全体,CHD7は小脳,CHD8は前頭前皮質において高発現を示す.また,CHD8はWntシグナリング経路の主要因子であるβ-カテニンと結合し,その転写活性を阻害する.Wntシグナリングは脳神経発達にも不可欠であり,神経細胞前駆体への発現誘導や神経細胞の層構造配置に関わることが知られている.そのためASDではCHD8のゲノム変異によってCHD8機能が失われることで,Wntシグナリングの下流遺伝子を活性化して,さまざまな神経発達機構に影響を与えると考えられている.モデルマウス研究では,Chd8ハプロ不全*1マウス(Chd8変異をヘテロ接合で有するマウス)が,不安の増強,反復行動,社会的行動の変化など,ASD様の行動の特徴を現すことを示すことが確認された39).遺伝子発現解析から,Chd8ハプロ不全マウスの胚では早期胎性遺伝子の発現上昇,後期胎性遺伝子の発現減少が確認され,さらに胎仔の脳重量の増加が確認された.この研究ではChd8発現減少によるWntシグナリング関連遺伝子の発現変動は確認されなかったが,神経幹細胞やES細胞で神経マーカー遺伝子の転写抑制に働くREST(RE-1 silencing transcription factor)の活性化が確認された.RESTはWntシグナリング関連遺伝子の抑制にも関与していることが示唆されており,REST活性化がChd8発現減少によるWntシグナリングへの影響を相殺したのかもしれない40).また,RESTの活性化はASDのヒト脳組織でも観察されたため,CHD8ハプロ不全はRESTの異常活性による神経幹細胞の分化障害を及ぼしている可能性が示唆される.別グループの研究においても,Chd8ハプロ不全による皮質,海馬,扁桃体の体積増加が成体マウスで確認された41).胎仔マウスでも神経細胞前駆体の増加が確認されたため,Chd8ハプロ不全により胎仔期の遺伝子発現変動が神経細胞数の増加や成体の脳体積の増加に影響したと考えられる.さらに別グループの作製したChd8ヘテロマウスでは,成体の遺伝子発現解析において,神経シナプス接着分子に関する遺伝子群(Kank1, Cntnap5b, Cntn6, Ankrd11, Pcdh15, Pcdha8, Pcdha9)の発現変動が確認された42).クロマチンリモデリング遺伝子のゲノム変異が正常な神経発達に必要な遺伝子発現を変動させ,将来的にも神経機能やシナプス接続状態に関わる遺伝子発現に影響を与え,行動異常を生じさせている可能性が示唆された.
MECP2はX染色体に局在するレット症候群の原因遺伝子として知られており,レット症候群の患者は精神遅滞,自閉傾向,運動失調,てんかんなどの症状を示す43).レット症候群も自閉症と同じくこれまで広範性発達障害に分類されていたが,DSM-5ではASDと区別されている.コードされているMECP2タンパク質はDNAがメチル化された遺伝子のプロモーター領域に結合し,SWI/SNFクロマチンリモデリング複合体の構成因子Brmと相互作用し,ヒストン脱アセチル化酵素HDAC1や転写抑制因子mSin3Aを誘導して,その標的遺伝子の発現を抑制する.一方,転写活性化因子CREBを誘導して,標的遺伝子の転写を活性化することも報告されている.MECP2タンパク質はさまざまな転写因子と相互作用し,多くの遺伝子発現調節に関与している.マウス研究では,Bdnf, Sst, Oprk1, Gamt, Gprin1が転写活性化,Mef2c, A2bp1が転写抑制化のMECP2標的遺伝子として示されている44).このうち転写抑制化の標的遺伝子であるMEF2CとA2BP1はどちらもASD患者でゲノム変異が見つかっており,ASD関連遺伝子として示唆されている遺伝子である.MECP2のゲノム変異を有することにより,標的となるASD関連遺伝子の発現変動を及ぼすことと,MEF2CやA2BP1のゲノム変異を有することは結果的にどちらもASD発症リスクを高めることにつながる.また,レット症候群のモデルマウスとしてMecp2ノックアウト(KO)マウスが作製され,多くの研究報告がなされている.Mecp2 KOマウスでは脳体積が減少しており,神経細胞の細胞体サイズも減少している45).これらの形態学的特徴はレット症候群患者の死後脳でも同様に観察されている.Mecp2 KOマウスでは,海馬初代培養細胞の興奮性シナプス数や興奮性入力頻度および振幅が減少し,皮質の錐体細胞においては,発火頻度の減少,興奮性入力の振幅減少が確認されている46, 47).このようにMecp2を欠失することは,遺伝子発現変動を引き起こすことで,結果的に神経生理機能にも大きな影響を及ぼす.また,細胞特異的に遺伝子組換えを生じさせるコンディショナルノックアウト(cKO)マウスの研究では,Mecp2欠損の影響は脳内において細胞ごとに異なることが明らかとなってきた.興奮性神経cKOと抑制性神経cKOにおいてASD様の社会性行動異常が確認されたが,ドーパミン神経cKOとセロトニン神経cKOではASD様行動が確認されなかった48–50).今後の遺伝子発現解析では,細胞を識別できるシングルセルRNAシーケンサー等を用いることで,細胞ごとにMECP2欠損の影響を受ける分子ネットワークが明らかとなってくるだろう.
FMR1は脆弱X症候群の原因遺伝子である.精神発達遅滞,注意欠陥と多動性,自閉傾向がみられ,身体的特徴として,長い顔,大きな耳,扁平足,関節の過伸展がみられる.脆弱X症候群患者におけるASD併存疾患の報告例は幅広く5~60%にわたる51).またASD患者のうち1~2%がFMR1のゲノム変異を持つと見積もられている.X染色体に局在するFMR1は5′-UTRにCGGリピート配列を持つ.通常6から40のリピート数であるが,脆弱X症候群患者では59から200以上のリピート数に至る.200以上にリピート数が増加すると,CGGリピート配列が高メチル化状態となりFRM1の遺伝子発現が消失する.FMR1がコードしているFMPRタンパク質はmRNAの翻訳機構に対して抑制的作用を持つ.FMRPはCYFIP1と複合体を形成し,翻訳前のmRNAをトラップしているeIF4Eに結合することで翻訳開始を阻害している.mRNAとeIF4EがFMRP複合体から外れると,eIF4GとeIF4Aとの複合体を形成し,リボソームを誘導することで翻訳が促進される.また,FMRPは神経細胞骨格やシナプス分子をコードしている遺伝子のmRNA(MAP1B, ARC, CAMK2A, DLG4, SAPAP3など)を主な標的としている.さらにASD関連遺伝子のデータベースであるSFARIとAutDBに登録されている120遺伝子がFMRPの標的遺伝子として示唆されている52).ゲノム解析においてもFMRP標的遺伝子のゲノム変異がASDで多く検出されているため,FMRPを中心とした遺伝子ネットワークを理解することはASD発症メカニズムを追う上で非常に重要である.興奮性シナプスに局在する代謝型グルタミン酸受容体mGluRは,活動依存的にローカルトランスレーション(樹状突起やシナプスにおいてmRNAが翻訳されること)を活性化することが知られている.小脳や海馬ではmGluRがリガンドを受容することで,AMPA受容体のエンドサイトーシスに作用するタンパク質(MAP1BやAPPなど)の翻訳が促進され,シナプスのLTD*2(long term depression,長期抑制:シナプスの伝達効率が弱化されること)が誘導される53, 54).FMRPの存在下では,MAP1BやAPPの翻訳が抑えられているが,Fmr1 KOマウスでは,これらのタンパク質が恒常的に翻訳され,海馬のシナプス長期抑制が過剰に生じると考えられている55).また,Fmr1 KOマウスでは,プレパルス抑制障害や記憶学習試験の成績低下,未成熟なスパイン密度の増加が確認されており,FMRPはシナプス可塑性や機能維持に重要であると示唆される.さらに,mGluRの下流経路を抑制するためにmGluR5アンタゴニストを投与することで,Fmr1 KOマウスのプレパルス抑制障害や未成熟スパインの過剰形成が改善する56).神経活動に応じてFMRPの標的mRNAがシナプス内で適切に翻訳されることが,シナプス機能を正常な状態に保つ上で重要である.ASD患者でFMRP標的遺伝子のゲノム変異が多数検出されていることは,ASDの脳内ではシナプス機能障害が生じていることを表すのかもしれない.
結節性硬化症の患者は,全身のさまざまな組織で良性腫瘍を生じやすく,てんかん発作や知的障害を有する.原因遺伝子として知られているTSC1とTSC2は,タンパク質合成を促進するmTORC1シグナリングの抑制的調節因子hamartinとtuberinをそれぞれコードしており,これらの働きがゲノム変異で失われるとmTORC1シグナリングが過剰に活性化し,タンパク質合成が促進される.また,結節性硬化症患者の約3割はASDと診断されることから,ASD発症メカニズムにmTORC1シグナリングが関わっていると考えられている57).モデルマウス研究では,Tsc1とTsc2はホモ接合で欠失すると胎生致死となることが知られている.ヘテロKOマウスでは,両者ともてんかん発作の症状はみられないが,記憶学習の障害を有する.Tsc1, Tsc2ともにヘテロKOマウスは社会性行動異常を示し,さらにmTORC1抑制剤であるラパマイシン投与によって社会性行動異常が改善することが報告されている58).mTORC1シグナリングにおいてTSC1とTSC2の上流に位置する分子をコードするPTENはがん抑制遺伝子の一つとして知られている.PTENのゲノム変異もASDで高頻度に検出されており,特に大頭症を有するASD患者では約7%がPTEN変異を持つ59).SFARIでは遺伝的に最もASD発症リスクが高いカテゴリー1の遺伝子として登録されている.PTENはタンパク質機能として脂質脱リン酸化活性を持ち,PI3Kを標的にしている.PTENの活性が失われるとPI3K/Aktシグナル経路の下流にあるmTORC1が過剰活性化され,タンパク質合成が亢進する.Pten KOマウスでは,社会性行動異常,不安の亢進,てんかん,プレパルス抑制の異常がみられる.mTORC1抑制剤のラパマイシンでこれらの行動異常が緩和される.また,ヒトのゲノム変異ではPTENタンパク質の細胞内局在を核局在優位性や細胞質局在優位性に変化させるものが見つかっている.細胞質局在優位性のゲノム変異を模したノックインマウスでは,脳サイズの肥大化やミクログリア活性,社会的動機づけ行動の増加が確認された60).このマウスの脳内遺伝子発現解析では,ASD関連遺伝子が有意に変動しており,その多くがシナプス伝達と免疫反応の分子ネットワークに属するものであった61).これら翻訳制御の機能障害(主に抑制機能を持つタンパク質の不活性化による翻訳機構の過剰活性化)が神経基盤に関わる構成分子の過剰合成につながり,ASDの一つの特徴であるシナプス過形成を引き起こし,正常な神経ネットワーク形成を乱しているのかもしれない.ASDにおける翻訳制御機構の標的分子量の増減を具体的に調べ,分子ネットワークの動態を明らかにすることが,ASD発症メカニズムを理解するために必要である.今後のさらなる研究が期待される.
ASD患者の死後脳研究においてスパイン異常や遺伝子発現解析におけるシナプス関連遺伝子の発現変動が多く検出されていることから,シナプス分子の機能異常とASD表現型が深く関連していると想定される.シナプス関連遺伝子のゲノム変異の多くはレアバリアントとして知られており,脳機能へ与える影響が大きい.SHANKファミリー(SHANK1, SHANK2, SHANK3)はスパインの足場タンパク質であり,PSD構成分子やアクチン骨格分子やグルタミン酸受容体と相互作用し,スパイン機能に関わる重要な因子である.SHANKファミリーのドメイン構造は共通しており,ANKドメイン(PSD分子SHARPINや細胞骨格分子α-fodrinと結合),SH3ドメイン(プロリンを多く含むペプチドに結合),PDZドメイン(PSD分子SAPAP1やAMPA受容体と結合),Proline-richドメイン(PSD分子Homerやcortactinと結合),SAMドメイン(自己重合に作用)を有する.SHANKファミリー遺伝子の変異がASDに占める割合は全体の1%を超えるくらいと見積もられる62).その中でもSHNAK3はSFARIのカテゴリー1のASD関連遺伝子として登録されており,ASD発症メカニズムを探る上でも注目すべき対象である.また,SHNAK3が局在する22番染色体の微小欠失による遺伝性疾患であるフェラン・マクダーミド症候群は,重度の言語障害や軽度の精神遅滞などを示し,0.5~2%ほどがASDと診断される63).SHANK3のゲノム変異は複数のエクソンで確認されており,タンパク質のドメイン機能に影響するゲノム変異はエクソンによって異なる9, 64–66).また,SHANK3変異のモデルマウス研究では,ANKドメインを含むエクソン4~9欠失,PDZドメインを含むエクソン13~16欠失,Proline-richドメインを含むエクソン21のナンセンス変異の各モデルにおいて,スパイン密度の減少や社会性行動異常が確認されている11, 67–70).一方で,ANKドメインの一部を含むエクソン9単独の欠失では電気生理学的には海馬と前頭前皮質の抑制性入力は減少しているが,社会性行動異常は確認されておらず,行動学的な表現型として表出されにくい71).また,SHANK2に限定したゲノムシーケンス研究では,1000人近くの被験者のうち約10%がSHANK2のミスセンス変異を有しており,さらにASD群3%,コントロール群1.5%の被験者がSHANKファミリー遺伝子の保存配列(タンパク質機能に関わり,種を超えて共通に保存されているDNA配列)に変異を持っていた72).これらのミスセンス変異を再現したShank2遺伝子をラット海馬初代培養細胞に過剰発現させると,ASD特異的なShank2変異の過剰発現細胞は樹状突起のシナプス密度を減少させたが,コントロール群特異的なShank2変異の過剰発現細胞はシナプス密度を変化させなかった.ASD症状のスペクトラム構造の複雑さを議論する上でも,欠失や重複による遺伝子発現量の増減だけでなく,ASD特有のゲノム変異よるタンパク質のドメイン機能異常を明確に調査することが重要である.
NRXNファミリー遺伝子(NRXN1, NRXN2, NRXN3)とNLNGファミリー遺伝子(NLGN1, NLGN2, NLGN3, NLGN4)はそれぞれプレシナプス,ポストシナプスに局在する接着分子をコードしており,タンパク質NRXNとNLGNは細胞外にN末端を突出させ,トランスシナプスに複合体を形成する.NLGNのC末端側では,PSD構成分子と結合し,NMDA受容体やAMPA受容体の膜局在に関わるので,これらの分子はシナプス形成もさることながらシナプス機能維持に重要であることが知られている.ゲノムワイドな遺伝学研究が始まる前の2003年に,ASD患者におけるNLGN3のR451Cミスセンス変異とNLGN4の一塩基挿入によるナンセンス変異が報告されたことから,これら接着分子のゲノム変異がASD発症メカニズムの主なターゲットとして注目を集めてきた.2007年と2008年には,NLGN3 R451CとNLGN4欠失のモデルマウスが作製され,両者とも行動学的に社会性行動異常などのASD症状を示すことが報告された12).しかしSHANKファミリーとは異なり,NLGNファミリーはゲノム変異が報告されているASD患者数は少ないが,NLGN変異はレアバリアントの一つとして知られている.NLGN3とNLGN4に比べて,NLGN1とNLGN2のASDケース報告はさらに少ない.近年NLGN1のP89Lミスセンス変異がASD家系で見つかり,Nlgn1 P89LノックインマウスにおけるASD様の行動異常も確認された73).また,NLGN1 P89Lは野生型NLGN1と比べてシナプス膜局在が低下することから,シナプス機能にも影響を与えていると考えられる.NLGN2はNLGNファミリーの中でも抑制性ポストシナプスに限定的に局在している分子であり,Nlgn2ノックアウトマウスでは抑制性シナプス数の減少,不安行動の増加や痛み感受性の低下が確認されているが,ASD症状はみられない.一方,NLGN3は主に興奮性シナプスに局在している分子であるが,Nlgn3 R451Cノックインマウスでは,脳内NLGN3タンパク質発現量が低下しているが,大脳皮質において興奮性入力は正常であり,逆に抑制性入力が増加している12).NGLNファミリーのゲノム変異の影響を知るためには今後の研究発展が期待される.
NRXNファミリーの中でもNRXN1は高頻度にゲノム変異が報告されている74, 75).これまでにホモ接合のNRXN1変異を持つASD患者は見つかっておらず,NRXN1はハプロ不全を及ぼす遺伝子として考えられている.しかしながら,モデルマウス研究ではNRXNファミリー遺伝子のゲノム変異はASD症状に対する影響力が小さいことが示されている.Nrnn1欠失モデルマウスでは,海馬における興奮性入力頻度の減少,プレパルス抑制の減少,グルーミング行動の増加,巣を作る活動量の低下,運動学習成績の上昇が確認されたが,社会性行動などの主要なASD様行動異常は示さなかった76).NRXN1の単一遺伝子変異では,電気生理学レベルではシナプス小胞放出能に異常が検出されるが,行動学レベルでは表現型が検出されにくいのかもしれない.また,NRXNファミリーは1000以上のスプライシングバリアントが確認されており,主に二つのプロモーター(αおよびβ)を持ち,細胞外領域に突出するN末端が長いタイプのα-NRXNと短いタイプのβ-NRXNがシナプス上に存在している.NRXN1, NRXN2, NRXN3のα-NRXNはプレシナプスにおいてカルシウムチャネルと結合しており,α-NRXNのトリプルKOマウスではグルタミン酸の小胞放出能が低下する77).一方,Nrxn2単一のα-NRXN KOマウスでは小胞放出能の低下は限定的である.Nrxn1欠失モデルやNrxn2単一のα-NRXN KOマウスでは表現型が弱いことから,NRXNファミリー遺伝子のいずれかにゲノム変異が生じても,残りの遺伝子が正常であれば,それらが補償的に作用してゲノム変異の影響を抑えているのかもしれない.
これまでシナプス構造や電気生理学的なシナプス機能の解析には,主に脳組織の固定標本や生きた実験動物から取り出された脳スライスなどが用いられてきた.しかしながら,ASDにおけるシナプスの構造的,生理学的機能変化が脳全体に派生するようなネットワーク機能異常を調べるためには,新しい技術が必要となる.モデル動物研究では,二光子顕微鏡や径頭蓋カルシウムイメージングなどの光学技術を用いることで,生きた動物の神経活動状態を可視化することができる.ASDのモデル動物を用いた生体内イメージング研究はまだ少ないが,そのいくつかを紹介する.
生体内光学イメージングを行うためには,蛍光標識色素や蛍光タンパク質発現ベクターを用いて神経細胞を蛍光標識する.また,神経細胞に蛍光タンパク質を発現させるトランスジェニックマウスも用いられる.皮質深部まで可視化できる二光子顕微鏡では,蛍光標識した神経細胞を時系列で比較することで,樹状突起やスパインの形態変化を調べることができる.Fmr1 KOマウスの研究では,大脳皮質の体性感覚野,運動野,視覚野において錐体細胞のスパインターンオーバー(スパインの形成と除去の割合)の増加が報告されている78).また,Mecp2 KOマウスでは,体性感覚皮質における視床皮質軸索終末数の減少,spiny satellite neuronにおける樹状突起の分岐形成やスパイン密度の低下,スパインターンオーバーの増加が報告されている78).15q11-13重複マウスでは,発達期における皮質錐体細胞のスパイン密度に変化はないが,スパインターンオーバーは増加している18).また,15q11-13重複マウスの研究では,スパインを機能分類して解析しており,興奮性シナプスマーカーをもつスパインではスパインターンオーバーが増加しているが,抑制性シナプスマーカーをもつスパインでは変化がないことが確認された18).さらに,Nlgn3 R451Cノックインマウスにおいてもスパインターンオーバー異常が確認されている18).このように生体内イメージングによって生きた動物の脳内の神経細胞構造を可視化することで,固定標本では観察できなかった時系列変化を捉えることができ,ASDモデルに共通したスパインターンオーバーの増加という表現型が見いだされた(図3).
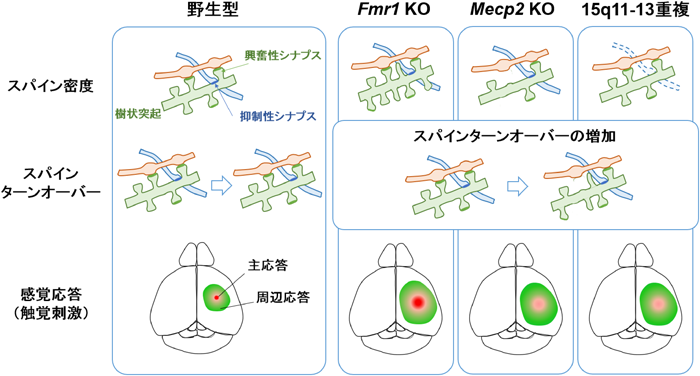
これまでにも解剖学的知見によるスパイン密度の増減がASD患者の死後脳やASDモデルマウスで報告されてきた.スパイン密度に関する研究では,ASDモデルマウスのうち,Fmr1 KOマウスではスパイン密度が野生型マウスと比べて増加(もしくは増減なし),Mecp2 KOマウスでは減少,15q11-13重複マウスでは増減がないと報告されている.しかしながら二光子顕微鏡による生体内イメージング研究では,いずれのマウスでも共通した表現型として,スパインターンオーバーの増加が報告されている.また,カルシウムイメージングや膜電位イメージングの神経機能解析では,ひげ刺激などの感覚刺激に対する応答異常が見つかっている.Fmr1 KOマウスでは応答が皮質領野間に広がる速度が増加しており,Mecp2 KOマウスと15q11-13重複マウスでは主応答の振幅が低下し,周辺応答エリアが拡大している.ゲノム変異による個々の分子メカニズムに加え,神経機能の共通した表現型をミクロ視点・マクロ視点の双方向から調べることで,脳基盤としてのASD発症メカニズムが明らかとなってくる.
生体内イメージングは形態だけではなく,神経活動を可視化するためにも有用なツールである.神経活動を可視化する方法として,神経発火時に生じる膜電位変化やカルシウムイオン流入を利用したカルシウムイメージングや膜電位イメージングがある.これらは,顕微鏡の解像度に応じて,細胞1個1個の神経活動から皮質全体にわたる広範囲な神経活動まで可視化することができる.触覚や視覚などの感覚入力は視床を通じて大脳皮質に送られる.上記生体内イメージングでは感覚刺激に対する受容野の皮質応答をミリ秒~秒スケールで観測することができる.膜電位イメージングを用いた研究では,Fmr1 KOマウスの触覚刺激に対する体性感覚皮質応答の伝播速度が亢進していることが報告された78).一方,Mecp2 KOマウスでは触覚刺激に対する応答が減弱していることが報告されている78).また,15q11-13重複マウスでは径頭蓋カルシウムイメージングによって,ひげ刺激に対する体性感覚皮質応答の減弱および受容野のシグナルノイズ比が低下していることが明らかとなった17).このように神経活動を観測した生体内イメージングによってASDモデルにおける脳内の感覚応答異常が確認されたことは,ASDの診断基準に含まれる感覚過敏や感覚鈍感といった表現型の脳内基盤を解明するための手がかりとなる(図3).
15q11-13重複マウスでは脳内セロトニン量が減少している16).その原因として,セロトニン神経が局在する縫線核の活動が低下していることが生体内イメージングの一つであるPETイメージングによって示唆された(図4)17).セロトニン神経の投射先経路に含まれる体性感覚皮質においても,上述のように径頭蓋カルシウムイメージングによって感覚応答のシグナルノイズ比低下が確認された.このような感覚応答異常は皮質ネットワークの抑制機能の脆弱性によって引き起こされることが知られている.15q11-13重複マウスでは体性感覚皮質錐体細胞における抑制性シナプス接続および抑制性シナプス入力が減少していることが確認された17).そのため15q11-13重複マウスでは感覚応答異常の原因として抑制機能の脆弱性が関与していることが示唆された.そして,15q11-13重複マウスの皮質抑制機能は,発達期にセロトニンを薬理学的に補強することで改善されることが明らかとなった17).さらに,この発達期セロトニン介入は15q11-13重複マウスの社会性行動を改善させた.このことから,15q11-13重複をもつASD発症メカニズムとして,脳内セロトニン減少が起因となる皮質抑制機能の低下,それに伴う感覚応答異常,そして正常な脳機能発達が損なわれることによって社会性行動異常につながることが示された(図4).このようにASDモデル動物を用いた研究では,ゲノム異常から脳機能までの一連の流れを調べ,その主要因となる原因分子を標的化することができるため,症状を緩和させる薬剤やASD発症を抑える薬剤開発につながる.臨床研究でもASD患者におけるセロトニン合成能の低下やセロトニントランスポーター結合能の低下が報告されていることから,セロトニン系の機能低下とASD発症には強い関係性が存在すると考えられている.将来的にもPETイメージングでヒトのセロトニン神経の機能低下を指標にするといった非侵襲的な脳機能イメージングがASD診断やASD発症リスクの予測に応用されることが期待される.
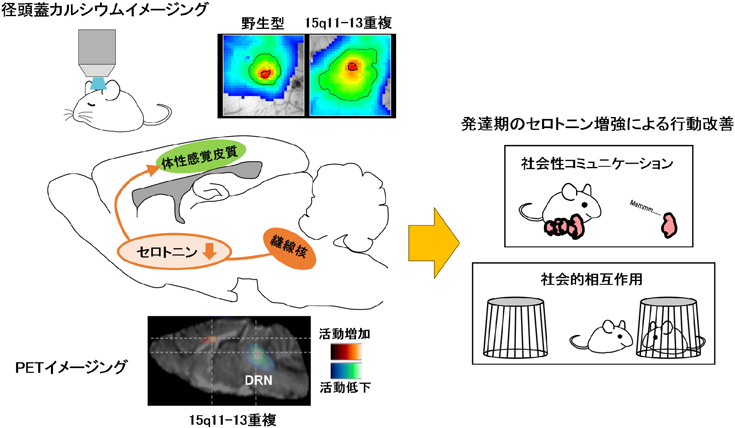
経頭蓋カルシウムイメージングは頭蓋越しにイメージングを行うため脳組織に対して非侵襲的な実験手法である.右側の画像は,麻酔下のマウスに対するひげ刺激時の皮質カルシウム反応を擬似カラーで表示したもの.15q11-13重複モデルマウスでは応答エリアが拡大していることがわかる.PETイメージングは放射性同位体を含む薬剤を被検体に投与し,その体内分布を画像化する非侵襲的な実験手法である.本画像は覚醒下のマウスに18F-FDG(フルデオキシグルコース)を注入し,脳内のグルコース代謝を擬似カラーで表したものである.15q11-13重複モデルマウスでは縫線核(DRN)の活動が低下していることが確認された.これらの知見から発達期のセロトニンに注目し,フルオキセチン(セロトニン再取り込み阻害剤)による薬理学的レスキューを行った結果,母仔間コミュニケーションや社会的相互作用といった社会性行動異常がフルオキセチン投与群の15q11-13重複モデルマウスで改善した.
ゲノム解析から見つかったASDの遺伝的変異は,レアバリアントやコモンバリアントを含め非常に多様性に富む.その中でもASD特異的に検出されやすいゲノム変異を集めると,共通性の高い分子ネットワークが現れてきた.主要なものは,「シナプス」と「クロマチンリモデリングおよび転写制御」である.また,遺伝子発現解析では,「シナプス」に加えて,「免疫・炎症」の分子ネットワークも現れてきた.どちらも共通点は「シナプス」であり,ゲノム変異もしくは遺伝子発現変動によってシナプス機能の変化が脳機能に影響を与えるのは明らかである.次に重要な問いは,ASD患者ではシナプスがどのように変化し,どのように機能障害を有しているのか?である.しかし,生体脳においてシナプス機能を調べることは難しい.これまでに死後脳検体から皮質錐体細胞のスパインを解析した研究では,ASD検体ではコントロール検体に比べて錐体細胞のスパイン密度が増加しており,スパイン密度の増加は脳重量が小さい人ほど顕著な傾向にあることが報告された79).さらに,情動と関連づけられている脳領域の扁桃体における死後脳研究では,ASD検体の扁桃体のスパイン密度が18歳未満の子供の脳で増加しているが,18歳以上の大人の脳では差がないことが報告されている80).そのため,発達期のスパイン密度が増加することが,神経回路の正常な接続を阻害し,将来にわたってASD症状を引き起こす原因の一つと考えられている.しかし,前節で述べたようにシナプス分子SHANKやNRXNのゲノム変異モデルマウス研究ではスパイン密度の減少やシナプス機能の低下が報告されている.一方でFmr1 KOマウスはスパイン密度が増加しており,15q11-13重複マウスはスパイン密度が野生型マウスと変わらない18, 81).スパイン密度の表現型が異なるにも関わらず,Fmr1ノックアウトマウス,15q11-13重複マウス,Nlgn3 R451Cノックインマウスでは,スパインのターンオーバー頻度が増加しているといった共通点を持つ18, 81).ASDにおけるスパイン密度の増減は,ある一時的な情報であり,多くても少なくてもASDの表現型に影響すると考えられる.スパインやシナプスによって表される神経どうしの接続状態が機能的に維持されているか否かがASD発症につながるより重要な表現型であるだろう.前節で述べたように生体内における機能解析が進むことでASD表現型の新しい指標が明らかとなることが期待される.
ASDのゲノム変異はシナプス分子に直接影響するだけではなく,遺伝子発現システム全体に影響を及ぼす.そのため,特に発達期における脳内の遺伝子発現パターンが正常の範疇から外れることで,シナプス形成や神経機能の正常発達が阻害されると考えられている.ASD発症リスクを抑えるためには,ゲノム変異の種類によらず,神経細胞における発達時の正常な遺伝子発現パターンを誘導しなくてはならない.それが可能になることで,正常な神経機能や接続状態を維持し,脳機能を正常に発達させることができる.ゲノム変異による分子ネットワーク変動の影響を発達期において時空間的に解析することが,今後のASD研究の発展につながるだろう.
1) Bourgeron, T. (2016) Current knowledge on the genetics of autism and propositions for future research. C. R. Biol., 339, 300–307.
2) Liu, X. & Takumi, T. (2014) Genomic and genetic aspects of autism spectrum disorder. Biochem. Biophys. Res. Commun., 452, 244–253.
3) Vorstman, J.a.S., Staal, W.G., van Daalen, E., van Engeland, H., Hochstenbach, P.F.R., & Franke, L. (2006) Identification of novel autism candidate regions through analysis of reported cytogenetic abnormalities associated with autism. Mol. Psychiatry, 11, 18–28.
4) Levy, D., Ronemus, M., Yamrom, B., Lee, Y., Leotta, A., Kendall, J., Marks, S., Lakshmi, B., Pai, D., Ye, K., et al. (2011) Rare de novo and transmitted copy-number variation in autistic spectrum disorders. Neuron, 70, 886–897.
5) Sanders, S.J., Ercan-Sencicek, A.G., Hus, V., Luo, R., Murtha, M.T., Moreno-De-Luca, D., Chu, S.H., Moreau, M.P., Gupta, A.R., Thomson, S.A., et al. (2011) Multiple recurrent de novo CNVs, including duplications of the 7q11.23 Williams syndrome region, are strongly associated with autism. Neuron, 70, 863–885.
6) Takumi, T. & Tamada, K. (2018) CNV biology in neurodevelopmental disorders. Curr. Opin. Neurobiol., 48, 183–192.
7) Kemper, T.L. & Bauman, M. (1998) Neuropathology of infantile autism. J. Neuropathol. Exp. Neurol., 57, 645–652.
8) Kaizuka, T. & Takumi, T. (2018) Postsynaptic density proteins and their involvement in neurodevelopmental disorders. J. Biochem., 163, 447–455.
9) Durand, C.M., Betancur, C., Boeckers, T.M., Bockmann, J., Chaste, P., Fauchereau, F., Nygren, G., Rastam, M., Gillberg, I.C., Anckarsäter, H., et al. (2007) Mutations in the gene encoding the synaptic scaffolding protein SHANK3 are associated with autism spectrum disorders. Nat. Genet., 39, 25–27.
10) Jamain, S., Quach, H., Betancur, C., Råstam, M., Colineaux, C., Gillberg, I.C., Soderstrom, H., Giros, B., Leboyer, M., Gillberg, C., et al.; Paris Autism Research International Sibpair Study. (2003) Mutations of the X-linked genes encoding neuroligins NLGN3 and NLGN4 are associated with autism. Nat. Genet., 34, 27–29.
11) Peça, J., Feliciano, C., Ting, J.T., Wang, W., Wells, M.F., Venkatraman, T.N., Lascola, C.D., Fu, Z., & Feng, G. (2011) Shank3 mutant mice display autistic-like behaviours and striatal dysfunction. Nature, 472, 437–442.
12) Tabuchi, K., Blundell, J., Etherton, M.R., Hammer, R.E., Liu, X., Powell, C.M., & Südhof, T.C. (2007) A neuroligin-3 mutation implicated in autism increases inhibitory synaptic transmission in mice. Science, 318, 71–76.
13) Pinto, D., Delaby, E., Merico, D., Barbosa, M., Merikangas, A., Klei, L., Thiruvahindrapuram, B., Xu, X., Ziman, R., Wang, Z., et al. (2014) Convergence of genes and cellular pathways dysregulated in autism spectrum disorders. Am. J. Hum. Genet., 94, 677–694.
14) Horev, G., Ellegood, J., Lerch, J.P., Son, Y.-E.E., Muthuswamy, L., Vogel, H., Krieger, A.M., Buja, A., Henkelman, R.M., Wigler, M., et al. (2011) Dosage-dependent phenotypes in models of 16p11.2 lesions found in autism. Proc. Natl. Acad. Sci. USA, 108, 17076–17081.
15) Nakatani, J., Tamada, K., Hatanaka, F., Ise, S., Ohta, H., Inoue, K., Tomonaga, S., Watanabe, Y., Chung, Y.J., Banerjee, R., et al. (2009) Abnormal behavior in a chromosome-engineered mouse model for human 15q11-13 duplication seen in autism. Cell, 137, 1235–1246.
16) Tamada, K., Tomonaga, S., Hatanaka, F., Nakai, N., Takao, K., Miyakawa, T., Nakatani, J., & Takumi, T. (2010) Decreased exploratory activity in a mouse model of 15q duplication syndrome; Implications for disturbance of serotonin signaling. PLoS ONE, 5, e15126.
17) Nakai, N., Nagano, M., Saitow, F., Watanabe, Y., Kawamura, Y., Kawamoto, A., Tamada, K., Mizuma, H., Onoe, H., Watanabe, Y., et al. (2017) Serotonin rebalances cortical tuning and behavior linked to autism symptoms in 15q11-13 CNV mice. Sci. Adv., 3, e1603001.
18) Isshiki, M., Tanaka, S., Kuriu, T., Tabuchi, K., Takumi, T., & Okabe, S. (2014) Enhanced synapse remodelling as a common phenotype in mouse models of autism. Nat. Commun., 5, 4742.
19) Yang, M., Mahrt, E.J., Lewis, F., Foley, G., Portmann, T., Dolmetsch, R.E., Portfors, C.V., & Crawley, J.N. (2015) 16p11.2 deletion syndrome mice display sensory and ultrasonic vocalization deficits during social interactions. Autism Res. Off. J. Int. Soc. Autism Res., 8, 507–521.
20) Blumenthal, I., Ragavendran, A., Erdin, S., Klei, L., Sugathan, A., Guide, J.R., Manavalan, P., Zhou, J.Q., Wheeler, V.C., Levin, J.Z., et al. (2014) Transcriptional consequences of 16p11.2 deletion and duplication in mouse cortex and multiplex autism families. Am. J. Hum. Genet., 94, 870–883.
21) Iossifov, I., O’Roak, B.J., Sanders, S.J., Ronemus, M., Krumm, N., Levy, D., Stessman, H.A., Witherspoon, K.T., Vives, L., Patterson, K.E., et al. (2014) The contribution of de novo coding mutations to autism spectrum disorder. Nature, 515, 216–221.
22) Liu, L., Lei, J., Sanders, S.J., Willsey, A.J., Kou, Y., Cicek, A.E., Klei, L., Lu, C., He, X., Li, M., et al. (2014) DAWN: A framework to identify autism genes and subnetworks using gene expression and genetics. Mol. Autism, 5, 22.
23) Voineagu, I., Wang, X., Johnston, P., Lowe, J.K., Tian, Y., Horvath, S., Mill, J., Cantor, R.M., Blencowe, B.J., & Geschwind, D.H. (2011) Transcriptomic analysis of autistic brain reveals convergent molecular pathology. Nature, 474, 380–384.
24) Gupta, S., Ellis, S.E., Ashar, F.N., Moes, A., Bader, J.S., Zhan, J., West, A.B., & Arking, D.E. (2014) Transcriptome analysis reveals dysregulation of innate immune response genes and neuronal activity-dependent genes in autism. Nat. Commun., 5, 5748.
25) Nardone, S., Sams, D.S., Reuveni, E., Getselter, D., Oron, O., Karpuj, M., & Elliott, E. (2014) DNA methylation analysis of the autistic brain reveals multiple dysregulated biological pathways. Transl. Psychiatry, 4, e433.
26) Nardone, S., Sams, D.S., Zito, A., Reuveni, E., & Elliott, E. (2017) Dysregulation of cortical neuron DNA methylation profile in autism spectrum disorder. Cereb. Cortex N. Y. N., 1991, 5739–5754.
27) Ellis, S.E., Gupta, S., Moes, A., West, A.B., & Arking, D.E. (2017) Exaggerated CpH methylation in the autism-affected brain. Mol. Autism, 8, 6.
28) Ginsberg, M.R., Rubin, R.A., Falcone, T., Ting, A.H., & Natowicz, M.R. (2012) Brain transcriptional and epigenetic associations with autism. PLoS ONE, 7, e44736.
29) Wang, P., Zhao, D., Lachman, H.M., & Zheng, D. (2018) Enriched expression of genes associated with autism spectrum disorders in human inhibitory neurons. Transl. Psychiatry, 8, 13.
30) Grabrucker, A.M. (2012) Environmental factors in autism. Front. Psychiatry, 3, 118.
31) Modabbernia, A., Velthorst, E., & Reichenberg, A. (2017) Environmental risk factors for autism:an evidence-based review of systematic reviews and meta-analyses. Mol. Autism, 8, 13.
32) Richetto, J., Massart, R., Weber-Stadlbauer, U., Szyf, M., Riva, M.A., & Meyer, U. (2017) Genome-wide DNA methylation changes in a mouse model of infection-mediated neurodevelopmental disorders. Biol. Psychiatry, 81, 265–276.
33) Coogan, A. & O’Connor, J.J. (1997) Inhibition of NMDA receptor-mediated synaptic transmission in the rat dentate gyrus in vitro by IL-1 beta. Neuroreport, 8, 2107–2110.
34) Lai, A.Y., Swayze, R.D., El-Husseini, A., & Song, C. (2006) Interleukin-1 beta modulates AMPA receptor expression and phosphorylation in hippocampal neurons. J. Neuroimmunol., 175, 97–106.
35) Murray, C.A., McGahon, B., McBennett, S., & Lynch, M.A. (1997) Interleukin-1 beta inhibits glutamate release in hippocampus of young, but not aged, rats. Neurobiol. Aging, 18, 343–348.
36) Tancredi, V., D’Antuono, M., Cafè, C., Giovedì, S., Buè, M.C., D’Arcangelo, G., Onofri, F., & Benfenati, F. (2000) The inhibitory effects of interleukin-6 on synaptic plasticity in the rat hippocampus are associated with an inhibition of mitogen-activated protein kinase ERK. J. Neurochem., 75, 634–643.
37) Pickering, M., Cumiskey, D., & O’Connor, J.J. (2005) Actions of TNF-alpha on glutamatergic synaptic transmission in the central nervous system. Exp. Physiol., 90, 663–670.
38) Vargas, D.L., Nascimbene, C., Krishnan, C., Zimmerman, A.W., & Pardo, C.A. (2005) Neuroglial activation and neuroinflammation in the brain of patients with autism. Ann. Neurol., 57, 67–81.
39) Katayama, Y., Nishiyama, M., Shoji, H., Ohkawa, Y., Kawamura, A., Sato, T., Suyama, M., Takumi, T., Miyakawa, T., & Nakayama, K.I. (2016) CHD8 haploinsufficiency results in autistic-like phenotypes in mice. Nature, 537, 675–679.
40) Johnson, R., Teh, C.H., Kunarso, G., Wong, K.Y., Srinivasan, G., Cooper, M.L., Volta, M., Chan, S.S., Lipovich, L., Pollard, S.M., et al. (2008) REST regulates distinct transcriptional networks in embryonic and neural stem cells. PLoS Biol., 6, e256.
41) Gompers, A.L., Su-Feher, L., Ellegood, J., Copping, N.A., Riyadh, M.A., Stradleigh, T.W., Pride, M.C., Schaffler, M.D., Wade, A.A., Catta-Preta, R., et al. (2017) Germline Chd8 haploinsufficiency alters brain development in mouse. Nat. Neurosci., 20, 1062–1073.
42) Platt, R.J., Zhou, Y., Slaymaker, I.M., Shetty, A.S., Weisbach, N.R., Kim, J.-A., Sharma, J., Desai, M., Sood, S., Kempton, H.R., et al. (2017) Chd8 mutation leads to autistic-like behaviors and impaired striatal circuits. Cell Reports, 19, 335–350.
43) Hagberg, B., Aicardi, J., Dias, K., & Ramos, O. (1983) A progressive syndrome of autism, dementia, ataxia, and loss of purposeful hand use in girls: Rett’s syndrome:report of 35 cases. Ann. Neurol., 14, 471–479.
44) Chahrour, M., Jung, S.Y., Shaw, C., Zhou, X., Wong, S.T.C., Qin, J., & Zoghbi, H.Y. (2008) MeCP2, a key contributor to neurological disease, activates and represses transcription. Science, 320, 1224–1229.
45) Chen, R.Z., Akbarian, S., Tudor, M., & Jaenisch, R. (2001) Deficiency of methyl-CpG binding protein-2 in CNS neurons results in a Rett-like phenotype in mice. Nat. Genet., 27, 327–331.
46) Chao, H.-T., Zoghbi, H.Y., & Rosenmund, C. (2007) MeCP2 controls excitatory synaptic strength by regulating glutamatergic synapse number. Neuron, 56, 58–65.
47) Dani, V.S., Chang, Q., Maffei, A., Turrigiano, G.G., Jaenisch, R., & Nelson, S.B. (2005) Reduced cortical activity due to a shift in the balance between excitation and inhibition in a mouse model of Rett syndrome. Proc. Natl. Acad. Sci. USA, 102, 12560–12565.
48) Chao, H.-T., Chen, H., Samaco, R.C., Xue, M., Chahrour, M., Yoo, J., Neul, J.L., Gong, S., Lu, H.-C., Heintz, N., et al. (2010) Dysfunction in GABA signalling mediates autism-like stereotypies and Rett syndrome phenotypes. Nature, 468, 263–269.
49) Gemelli, T., Berton, O., Nelson, E.D., Perrotti, L.I., Jaenisch, R., & Monteggia, L.M. (2006) Postnatal loss of methyl-CpG binding protein 2 in the forebrain is sufficient to mediate behavioral aspects of Rett syndrome in mice. Biol. Psychiatry, 59, 468–476.
50) Samaco, R.C., Mandel-Brehm, C., Chao, H.-T., Ward, C.S., Fyffe-Maricich, S.L., Ren, J., Hyland, K., Thaller, C., Maricich, S.M., Humphreys, P., et al. (2009) Loss of MeCP2 in aminergic neurons causes cell-autonomous defects in neurotransmitter synthesis and specific behavioral abnormalities. Proc. Natl. Acad. Sci. USA, 106, 21966–21971.
51) Devitt, N.M., Gallagher, L., & Reilly, R.B. (2015) Autism Spectrum Disorder (ASD) and Fragile X Syndrome (FXS): Two overlapping disorders reviewed through electroencephalography-what can be interpreted from the available information? Brain Sci., 5, 92–117.
52) Fernández, E., Rajan, N., & Bagni, C. (2013) The FMRP regulon:from targets to disease convergence. Front. Neurosci., 7, 191.
53) Benoist, M., Palenzuela, R., Rozas, C., Rojas, P., Tortosa, E., Morales, B., González-Billault, C., Ávila, J., & Esteban, J.A. (2013) MAP1B-dependent Rac activation is required for AMPA receptor endocytosis during long-term depression. EMBO J., 32, 2287–2299.
54) Pasciuto, E., Ahmed, T., Wahle, T., Gardoni, F., D’Andrea, L., Pacini, L., Jacquemont, S., Tassone, F., Balschun, D., Dotti, C.G., et al. (2015) Dysregulated ADAM10-mediated processing of APP during a critical time window leads to synaptic deficits in fragile X syndrome. Neuron, 87, 382–398.
55) Lüscher, C. & Huber, K.M. (2010) Group 1 mGluR-dependent synaptic long-term depression:mechanisms and implications for circuitry and disease. Neuron, 65, 445–459.
56) de Vrij, F.M.S., Levenga, J., van der Linde, H.C., Koekkoek, S.K., De Zeeuw, C.I., Nelson, D.L., Oostra, B.A., & Willemsen, R. (2008) Rescue of behavioral phenotype and neuronal protrusion morphology in Fmr1 KO mice. Neurobiol. Dis., 31, 127–132.
57) Gipson, T.T., Gerner, G., Wilson, M.A., Blue, M.E., & Johnston, M.V. (2013) Potential for treatment of severe autism in tuberous sclerosis complex. World J. Clin. Pediatr., 2, 16–25.
58) Sato, A., Kasai, S., Kobayashi, T., Takamatsu, Y., Hino, O., Ikeda, K., & Mizuguchi, M. (2012) Rapamycin reverses impaired social interaction in mouse models of tuberous sclerosis complex. Nat. Commun., 3, 1292.
59) Tilot, A.K., Frazier, T.W. 2nd, & Eng, C. (2015) Balancing proliferation and connectivity in PTEN-associated autism spectrum disorder. Neurother. J. Am. Soc. Exp. Neurother., 12, 609–619.
60) Tilot, A.K., Gaugler, M.K., Yu, Q., Romigh, T., Yu, W., Miller, R.H., Frazier, T.W. 2nd, & Eng, C. (2014) Germline disruption of Pten localization causes enhanced sex-dependent social motivation and increased glial production. Hum. Mol. Genet., 23, 3212–3227.
61) Tilot, A.K., Bebek, G., Niazi, F., Altemus, J.B., Romigh, T., Frazier, T.W., & Eng, C. (2016) Neural transcriptome of constitutional Pten dysfunction in mice and its relevance to human idiopathic autism spectrum disorder. Mol. Psychiatry, 21, 118–125.
62) Leblond, C.S., Nava, C., Polge, A., Gauthier, J., Huguet, G., Lumbroso, S., Giuliano, F., Stordeur, C., Depienne, C., Mouzat, K., et al. (2014) Meta-analysis of SHANK mutations in autism spectrum disorders: A gradient of severity in cognitive impairments. PLoS Genet., 10, e1004580.
63) Kolevzon, A., Angarita, B., Bush, L., Wang, A.T., Frank, Y., Yang, A., Rapaport, R., Saland, J., Srivastava, S., Farrell, C., et al. (2014) Phelan-McDermid syndrome: A review of the literature and practice parameters for medical assessment and monitoring. J. Neurodev. Disord., 6, 39.
64) Gauthier, J., Spiegelman, D., Piton, A., Lafrenière, R.G., Laurent, S., St-Onge, J., Lapointe, L., Hamdan, F.F., Cossette, P., Mottron, L., et al. (2009) Novel de novo SHANK3 mutation in autistic patients. Am. J. Med. Genet. Part B Neuropsychiatr. Genet. Off. Publ. Int. Soc. Psychiatr. Genet., 150B, 421–424.
65) Moessner, R., Marshall, C.R., Sutcliffe, J.S., Skaug, J., Pinto, D., Vincent, J., Zwaigenbaum, L., Fernandez, B., Roberts, W., Szatmari, P., et al. (2007) Contribution of SHANK3 mutations to autism spectrum disorder. Am. J. Hum. Genet., 81, 1289–1297.
66) Monteiro, P. & Feng, G. (2017) SHANK proteins: Roles at the synapse and in autism spectrum disorder. Nat. Rev. Neurosci., 18, 147–157.
67) Bozdagi, O., Sakurai, T., Papapetrou, D., Wang, X., Dickstein, D.L., Takahashi, N., Kajiwara, Y., Yang, M., Katz, A.M., Scattoni, M.L., et al. (2010) Haploinsufficiency of the autism-associated Shank3 gene leads to deficits in synaptic function, social interaction, and social communication. Mol. Autism, 1, 15.
68) Kouser, M., Speed, H.E., Dewey, C.M., Reimers, J.M., Widman, A.J., Gupta, N., Liu, S., Jaramillo, T.C., Bangash, M., Xiao, B., et al. (2013) Loss of predominant Shank3 isoforms results in hippocampus-dependent impairments in behavior and synaptic transmission. J. Neurosci., 33, 18448–18468.
69) Mei, Y., Monteiro, P., Zhou, Y., Kim, J.-A., Gao, X., Fu, Z., & Feng, G. (2016) Adult restoration of Shank3 expression rescues selective autistic-like phenotypes. Nature, 530, 481–484.
70) Zhou, Y., Kaiser, T., Monteiro, P., Zhang, X., Van der Goes, M.S., Wang, D., Barak, B., Zeng, M., Li, C., Lu, C., et al. (2016) Mice with Shank3 mutations associated with ASD and schizophrenia display both shared and distinct defects. Neuron, 89, 147–162.
71) Lee, J., Chung, C., Ha, S., Lee, D., Kim, D.-Y., Kim, H., & Kim, E. (2015) Shank3-mutant mice lacking exon 9 show altered excitation/inhibition balance, enhanced rearing, and spatial memory deficit. Front. Cell. Neurosci., 9, 94.
72) Leblond, C.S., Heinrich, J., Delorme, R., Proepper, C., Betancur, C., Huguet, G., Konyukh, M., Chaste, P., Ey, E., Rastam, M., et al. (2012) Genetic and functional analyses of SHANK2 mutations suggest a multiple hit model of autism spectrum disorders. PLoS Genet., 8, e1002521.
73) Nakanishi, M., Nomura, J., Ji, X., Tamada, K., Arai, T., Takahashi, E., Bućan, M., & Takumi, T. (2017) Functional significance of rare neuroligin 1 variants found in autism. PLoS Genet., 13, e1006940.
74) Feng, J., Schroer, R., Yan, J., Song, W., Yang, C., Bockholt, A., Cook, E.H. Jr., Skinner, C., Schwartz, C.E., & Sommer, S.S. (2006) High frequency of neurexin 1beta signal peptide structural variants in patients with autism. Neurosci. Lett., 409, 10–13.
75) Kim, H.-G., Kishikawa, S., Higgins, A.W., Seong, I.-S., Donovan, D.J., Shen, Y., Lally, E., Weiss, L.A., Najm, J., Kutsche, K., et al. (2008) Disruption of neurexin 1 associated with autism spectrum disorder. Am. J. Hum. Genet., 82, 199–207.
76) Etherton, M.R., Blaiss, C.A., Powell, C.M., & Südhof, T.C. (2009) Mouse neurexin-1alpha deletion causes correlated electrophysiological and behavioral changes consistent with cognitive impairments. Proc. Natl. Acad. Sci. USA, 106, 17998–18003.
77) Missler, M., Zhang, W., Rohlmann, A., Kattenstroth, G., Hammer, R.E., Gottmann, K., & Südhof, T.C. (2003) Alpha-neurexins couple Ca2+ channels to synaptic vesicle exocytosis. Nature, 423, 939–948.
78) Nakai, N., Takumi, T., Nakai, J., & Sato, M. (2018) Common defects of spine dynamics and circuit function in neurodevelopmental disorders: A systematic review of findings from in vivo optical imaging of mouse models. Front. Neurosci., 12, 412.
79) Hutsler, J.J. & Zhang, H. (2010) Increased dendritic spine densities on cortical projection neurons in autism spectrum disorders. Brain Res., 1309, 83–94.
80) Weir, R.K., Bauman, M.D., Jacobs, B., & Schumann, C.M. (2018) Protracted dendritic growth in the typically developing human amygdala and increased spine density in young ASD brains. J. Comp. Neurol., 526, 262–274.
81) Cruz-Martín, A., Crespo, M., & Portera-Cailliau, C. (2010) Delayed stabilization of dendritic spines in fragile X mice. J. Neurosci., 30, 7793–7803.

理化学研究所研究員.生命科学博士.
意識とは? こころとは? 分子から神経回路までネットワークダイナミクスでアプローチする.
趣味酒場の探求,酒蔵の探求.

理化学研究所脳科学総合研究センターシニアチームリーダー.医学博士.
兵庫県生まれ.1990年京都大学大学院医学研究科修了.大阪バイオサイエンス研究所研究室長,広島大学医学部教授を経て,2013年より現職.
研究テーマと抱負自閉症を含む精神疾患の統合的研究,概日リズムの統合的研究,そしてその二つを結びつけること.
ウェブサイトhttp://takumi.brain.riken.jp
趣味ワインと阪神.
*1相同染色体のもつ一対の遺伝子のうちの片側に突然変異が起こっても,ほとんどの遺伝子では,もう片側の正常な遺伝子(ハプロイド)から作られるタンパク質で不足分をまかなえる.しかし,発生に関わるいくつかの重要な遺伝子においては片側だけではタンパク質量が不足することがあり,機能不全が生じる.このような現象をハプロ不全と呼ぶ.
*2個々のシナプスは可塑性を持つ.神経伝達物質の放出回数が高頻度になりシナプスの結びつきが強化されて伝達効率が上がることをLTP(長期増強),逆に低頻度になりシナプスの結びつきが弱化されて伝達効率が下がることをLTD(長期抑制)と呼ぶ.

This page was created on 2018-06-27T13:41:17.254+09:00
This page was last modified on 2018-08-17T09:29:57.219+09:00
このサイトは(株)国際文献社によって運用されています。