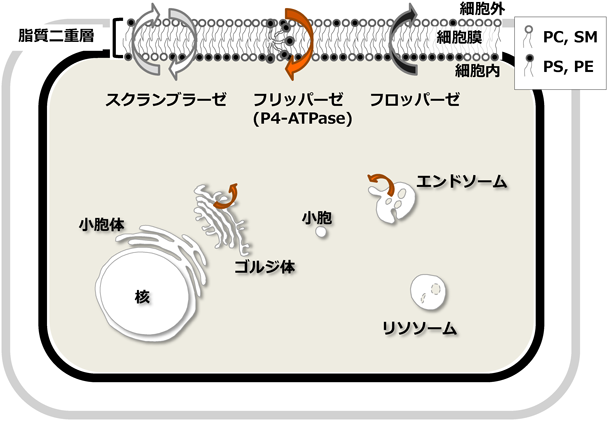
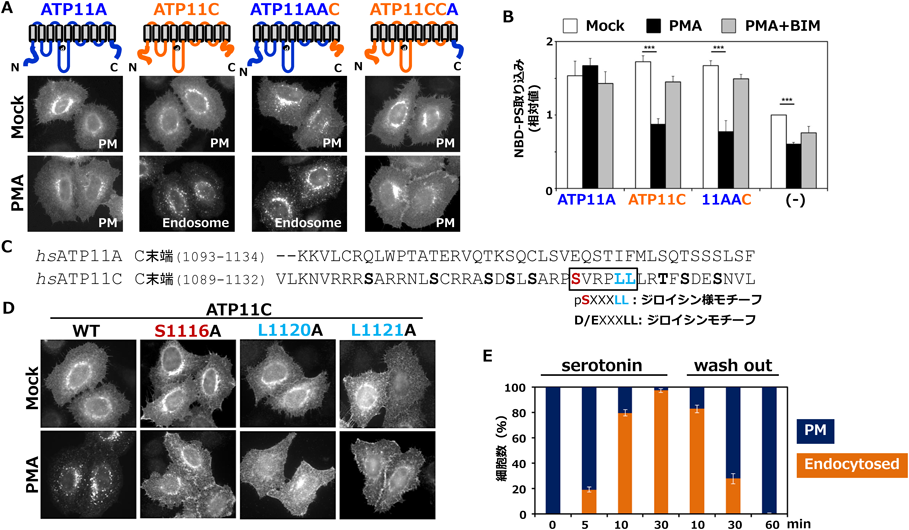
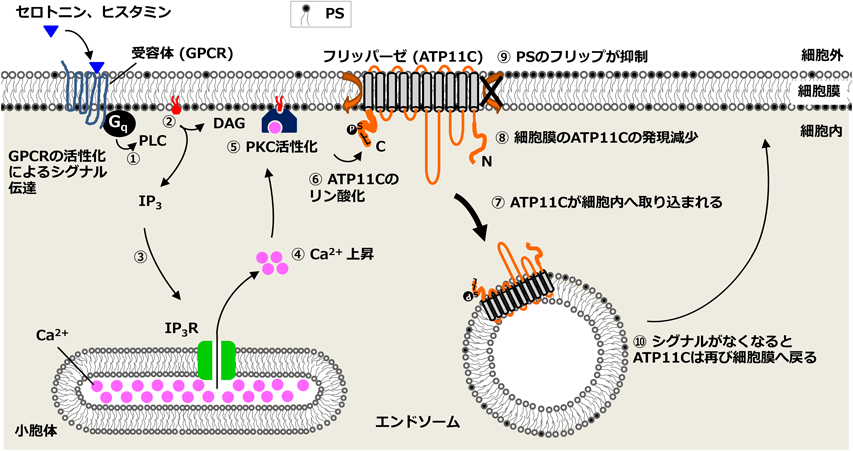
細胞膜ホスファチジルセリン–フリッパーゼの活性調節機構Regulation mechanism of the PS-flippase ATP11C at the plasma membrane
京都大学大学院薬学研究科Graduate School of Pharmaceutical Sciences, Kyoto University ◇ 〒606–8501 京都市左京区吉田下阿達町46–29 ◇ 46–29 Yoshida-shimo-adachi-cho, Sakyo-ku, Kyoto 606–8501, Japan
発行日:2018年8月25日Published: August 25, 2018