1)S1P受容体サブタイプとその発現
S1P1,S1P2,S1P3のmRNAは全身のほとんどすべての組織,臓器に発現し,一方,S1P4およびS1P5の発現はリンパ系組織や脳などの一部組織に限局している4, 5, 8, 9, 13).細胞レベルではS1P1は血管内皮に発現し,血管内皮に最も強発現するGPCRとされている14).S1P2とS1P3は血管内皮と平滑筋の両細胞に発現しているが,S1P1に比較して発現の程度は低い6, 7).S1P1,S1P2,S1P3の広範な臓器発現には,これら血管細胞の寄与がある.S1P1~S1P5の内因性リガンドは,S1Pの他に,dihydro-S1P(sphinganine 1-phosphate),phytoS1P(4-hydroxysphinganine 1-phosphate)があり,S1P1~S1P5はこれらのリガンドすべてに対してはnMレベルの親和性を有する.
2)S1P受容体のシグナル伝達機構
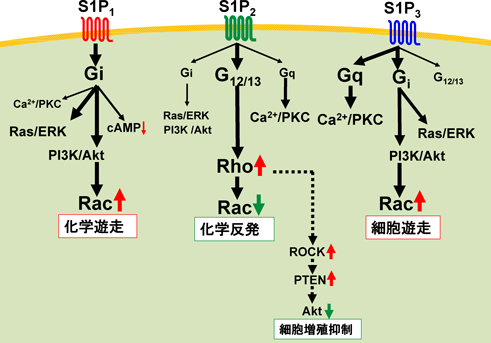
主要な受容体であるS1P1,S1P2,S1P3の各受容体について,筆者らを含むいくつかのグループによって詳しい検討がなされた(図2).S1P1は三量体Gタンパク質Giとのみ共役し,低分子量Gタンパク質Rasの活性化を介してERK(extracellular signal-regulated kinase)の活性化,ホスファチジルイノシトール3-キナーゼ(phosphoinositol 3-kinase:PI3K)を介してAktや低分子量Gタンパク質Racの活性化,ホスホリパーゼC(PLC)の活性化[Ca2+動員/Cキナーゼ(PKC)活性化],アデニル酸シクラーゼの抑制[サイクリックAMP(cAMP)産生の抑制]に共役している4, 5).特にRac活性化は,S1P1に特徴的な化学遊走(chemotaxis)反応の分子スイッチとして機能する15, 16).これらの反応はすべて細胞の百日咳毒素処理によって抑制される.一方,S1P2,S1P3の両受容体は複数種類の三量体Gタンパク質と共役しうる.しかし,受容体KOマウス胎仔由来の線維芽細胞(MEF)を用いた検討によると,S1P3は主としてGqに共役してPLCを活性化し,Ca2+動員とPKC活性化を引き起こし,S1P2はG12/13を介して低分子量Gタンパク質Rhoを活性化する17).S1P2はRhoの下流でRhoキナーゼ(ROCK/ROK)を活性化する.また,S1P2はRhoの下流でROCKを介さずにRacを抑制することにより化学反発(高濃度のS1Pを忌避する細胞遊走)を引き起こす15, 16).ROCKの下流では,3′特異的ホスホイノシチドホスファターゼであるphosphatase and tensin homolog(PTEN)を活性化し,ホスファチジルイノシトール3,4,5-トリスリン酸を減少させることによってAktを抑制する18, 19).この経路は,細胞増殖・生存の抑制や血管内皮におけるNO合成酵素eNOS活性抑制などを来す.
S1P受容体や代謝酵素SphK, SgpL遺伝子のノックアウトマウスの表現型,受容体のアゴニスト・アンタゴニストやSphK阻害薬の効果,S1Pトランスポーター遺伝子の解析などの結果に基づいて,S1Pの生体における主な機能は,1)血管の発生と機能の調節,2)白血液の体内動態の調節,3)神経系の発生,の三つにまとめられる.
1)血管作用
胎児期の血管は,脈管形成,血管新生(発芽[sprouting])そして血管成熟(壁細胞被覆)を経て発生し,さまざまな径の動脈および静脈ならびに毛細血管からなる血管系が構築される.S1P1-KO(全身)マウス胎仔では広汎な出血がみられE14.5日までに死亡した20).この時期の野生型マウスの大動脈では2~3層の平滑筋からなる中膜が形成されるが,KOマウスでは大動脈壁の背側部分でこの中膜平滑筋層を欠く結果,血管壁が脆弱となり出血した.血管内皮特異的コンディショナルS1P1-KOマウス胎仔が同様の異常な表現型を呈したことから,内皮S1P1の欠損が中膜平滑筋層の形成不全の原因とわかった21).血管新生部位では,発芽の最先端部位にtip cellと呼ばれる遊走活性の高い内皮細胞が存在し,その後方にはstalk cellと呼ばれる内皮細胞が並んで管腔を形成する.S1P1発現の詳細な解析により,S1P1は発芽部位のtip cell, stalk cellには発現があまりみられず,VEカドヘリン結合を介して安定化されている血管の非新生部位の内皮に強く発現していた22).内皮S1P1は内皮細胞間の結合[接着結合(adherens junction)]を強化して血管を安定化させ,内皮細胞のtip細胞化を阻害することにより発芽的血管新生を抑制することが複数の研究室からの報告で示された22, 23).当初報告されたS1P1-KO胎仔における血管平滑筋層の形成異常は過剰なtip cellの形成により壁細胞による被覆が障害された結果と考えられた.一方,S1P1とは異なるシグナル伝達を行うS1P2は,マウス未熟児網膜症モデルにおける低酸素に対する網膜血管新生を抑制することがS1P2-KOマウスの解析により最初に示された24).しかし,S1P2はこのモデルで硝子体内に侵入する病的血管新生をむしろ促進した.血管の平滑筋細胞はS1P2とS1P3を発現し,これら両受容体は平滑筋を収縮させ血管トーヌスを増加させる24).
2)白血球への作用
S1Pはリンパ球や単球/マクロファージなどの体内循環,活性化,分化を調節することにより,免疫,炎症応答において重要な役割をも果たす.特にTリンパ球に対する体内循環調節作用は顕著であり,この作用は主としてS1P1を介する.胸腺やリンパ節・腸管粘膜パイエル板などの二次リンパ組織の実質では,S1PはSpgLによって分解されるためにS1P濃度が血液・リンパ液に比較して低く保たれている.S1P1をノックアウトしたマウスでは,Tリンパ球は,S1P濃度の低い胸腺や二次リンパ組織の実質から,S1P濃度のはるかに高いリンパ液・血液中に遊出(egress)できないために,胸腺に存在する成熟途上のTリンパ球,二次リンパ組織内の成熟Tリンパ球数が増加し,血中や末梢組織にはTリンパ球がほとんど見いだされない25).S1P1は以下の三つのレベルでTリンパ球遊出を調節する.第一は,リンパ組織内と流出路(リンパ管や血管)の間のS1P濃度勾配の存在である.リンパ組織のS1P分解酵素SgpLの活性を阻害するとリンパ組織内のS1P濃度が上昇し濃度勾配が失われる結果,リンパ球のリンパ管への移行は阻害される.リンパ液中の高濃度S1Pの主要産生源はリンパ管内皮である.一方,胸腺では血管壁の神経堤由来の血管周囲細胞がS1Pを放出して血管周囲にリンパ球を引きつけ,一段とS1Pが高濃度である血管内への侵入を促す.第二は細胞表面受容体分子S1P1の発現レベルである.S1P1はS1Pの結合によって細胞内移行(internalization)し,低S1P濃度環境ではリサイクリングにより細胞表面S1P1発現レベルが回復する.この周期的な細胞表面におけるS1P1発現変動がリンパ球のリンパ組織通過時間に関与している可能性がある.実際に,S1P1細胞内移行の初発過程であるS1P1リン酸化を受けない変異S1P1ノックインマウスでは,S1P1細胞内移行が抑制されてS1P1アゴニスト投与後の血中リンパ球数の減少開始が遅延する.第三は,リンパ球において,S1P1遺伝子発現を促進する転写因子であるKruppel-like factor 2(KLF2)である.KLF2の発現が活性化リンパ球では低下する結果,S1P1遺伝子の発現低下が誘導される.
3)神経系の発生に及ぼす作用
胎仔の脳の神経細胞にS1P1の発現がみられ,SphK1およびSphK2のKOマウスでは脳の発生の著しい異常がみられた12).成獣マウスの脳組織では,神経細胞,グリア細胞の両者に複数のS1P受容体サブタイプが発現し,神経伝達への影響が報告された.
1)S1P2による血管バリア機能破綻の阻止
正常血管においては,細胞膜VEカドヘリン分子によって内皮細胞間に接着結合が形成され,この構造はVEカドヘリンの細胞内領域へのカテニンの結合により安定化・強化されている26).接着結合は血漿の血管外への漏洩を阻止する障壁として機能している.接着結合のバリア(障壁)機能が破綻すると血漿が血管外の組織間隙に漏洩して浮腫を生ずる.また,腫瘍内の新生血管ではバリア機能が低下し,これはがんの転移に関与する.さまざまな生理活性因子が接着結合の障壁機能に影響を及ぼす.活性化された血小板から放出される非ペプチド性のメディエーターがバリア機能を高めることは以前から知られていた.Garciaらは,血小板が放出するS1Pに着目してその内皮バリア機能に及ぼす作用を調べ,内皮単層培養においてS1Pがバリア機能を著しく高めることを見いだした26).S1Pとは逆に血液凝固に際して産生されるトロンビンは内皮バリア機能を傷害するが,S1Pはトロンビンによるバリア機能の低下を防いだ.このS1P作用は,S1P1によって引き起こされる内皮細胞間接着部位へのVEカドヘリンおよび安定化因子カテニンの集積促進を介した14, 26).S1Pのバリア増強作用の細胞内機構には,Gi-Rac経路,Rap経路が重要な役割を果たすことが示唆されている.
脂質メディエーターとしてのS1Pの特徴の一つは,上述のごとくS1Pが血漿にきわめて高濃度で存在する点である3).SphK1とSphK2の二重ノックアウト胎仔肝臓由来の造血細胞を移植したマウス(S1P-lessマウス)では,血漿S1P濃度は正常の1/10程度に低下した27).S1P-lessマウスでは,野生型マウスに比較して血管内皮細胞間の接着が弱まり,平常時においても血管透過性の亢進を示す.IgE/抗原感作によるアナフィラキシーモデル(受動型アナフィラキシーモデル)やアナフィラキシー誘発メディエーターであるヒスタミンや血小板活性化因子(PAF)の静脈内投与により血管透過性を亢進させると,S1P-lessマウスでは野生型マウスに比較して肺血管透過性の亢進が増強され,致死率が上昇した.S1P1アゴニストを投与すると,S1P-lessマウスで観察される血管透過性亢進と致死率悪化は改善した.このように,血漿S1Pは血管内皮細胞のS1P1を介して恒常的に血管透過性を低く保ち,さらにアナフィラキシー時の血管透過性亢進を抑制する.
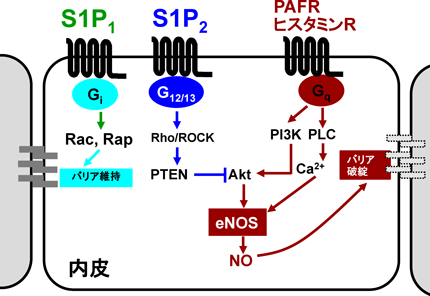
筆者らは,能動型アナフィラキシーモデル(ウシ血清アルブミンなどの異種タンパク質であらかじめ感作したマウスにこれらの抗原を再投与するモデル)を用いて,S1P2がバリア機能に及ぼす作用を検討した28).抗原再投与により,肥満細胞やマクロファージはIgE依存的にPAFやヒスタミンなどのアナフィラキシーメディエーターを大量に放出する.S1P2-KOマウスでは,血管透過性の基礎値には異常は認められないが,アナフィラキシー時の血管透過性亢進が顕著に悪化し死亡率が上昇した.同様の結果は他の研究者からも報告された29).また,S1P2-KOマウスではアナフィラキシーのメディエーターであるPAF投与による血管透過性亢進も悪化したことから,S1P2の作用点はPAF等のアナフィラキシーメディエーターの産生ではなく,これらメディエーターの内皮への作用(バリア機能の障害)の段階であることが示唆された.PAFやヒスタミンはリン酸化酵素Aktを介して内皮のeNOSを活性化し一酸化窒素(NO)産生を増強する30).産生されたNOはカテニンをニトロシル化してカテニン分解を促進する結果,細胞間接着装置であるVEカドヘリン–カテニン複合体が崩壊してバリア機能が破綻し血管透過性が亢進する.NO産生酵素eNOSのKOマウスではアナフィラキシーが著しく軽症化することから,バリア機能破綻においてはNOが重要な役割を果たすことが知られていた31).内皮におけるS1P2の作用機構を検討したところ,S1P2はPAFによるNO産生を抑制してカテニンのニトロシル化を抑制し,この結果VEカドヘリン−カテニン複合体の崩壊を防いだ.S1P2によるNO産生抑制は,eNOSを活性化するリン酸化酵素Aktの抑制によると考えられた.すなわち,S1P2の役割はアナフィラキシーなどのバリア破綻侵襲が加わったときにNO産生促進によって引き起こされる血管バリア破綻を,NO産生を抑制することによって防御することであった.一方,S1P1の役割はバリア機能を恒常的に維持することである.S1P1によるバリア機能の維持能力を超えたアナフィラキシーなどのバリア破綻侵襲が加わったときには,S1P2のバリア防御機能が重要となりバリア破綻を阻止する(図3).
血管内皮のバリア機能は,アナフィラキシーに限らず,局所の感染や非感染性の炎症,全身疾患である敗血症等の重症疾患に合併する急性肺水腫(acute respiratory distress syndrome:ARDS)などの疾患においても,破綻が生ずる.S1Pの血管バリア維持作用は,急性肺水腫やアナフィラキシーショックの血管透過性亢進に対する治療に応用できる可能性がある.急性肺水腫の動物モデルであるリポ多糖投与による肺水腫に対して,S1Pや体内でリン酸化を受けてS1P1アゴニストとして作用するFTY720の投与が肺水腫を軽減することが報告された32).筆者らは,S1P2-KOマウスではリポ多糖投与による血管バリア破綻が悪化することを見いだしており,S1P2はアナフィラキシーと同様にリポ多糖によるバリア破綻を防止すると考えている.肺水腫に対してFTY720以外のS1P1アゴニストの有効性も報告されている.しかし,これらのS1P1作動薬は反復投与により内皮S1P1の内在化を引き起こして逆にバリア機能低下をもたらすので,S1P1の内在化(脱感作)によるリバウンド現象は解決すべき重要な問題である.また,S1P2アゴニストも治療薬となる可能性がある.
2)S1P2による血管形成の抑制
血管内皮研究に用いられる培養内皮細胞の多くではS1P2の発現レベルは非常に低値あるいは検出感度以下であるため,in vitroでの血管内皮細胞におけるS1P2作用の報告はこれまで乏しかった.S1P2の血管新生における役割については,S1P2がマウス未熟児網膜症モデルにおける低酸素に対する網膜血管新生を抑制することがS1P2-KOマウスの解析により最初に示された33).しかし,このモデルでは,S1P2は硝子体内の病的な血管新生をむしろ促進した.
筆者らは,S1P2-KOマウスにおいて,S1P2が腫瘍血管新生を抑制することを見いだした19).ルイス肺がん,B16黒色腫をS1P2-KOおよび野生型マウスに同種移植すると,いずれの腫瘍の増殖も野生型マウスに比較してS1P2-KOマウスで亢進した.内皮マーカー抗体を用いた免疫染色により,S1P2-KOマウスでは野生型マウスに比較して,腫瘍内の新生血管密度が増加していた.また,S1P2-KOマウスでは腫瘍血管の平滑筋および周皮細胞の集積が亢進しており,血管の安定化・成熟が促進されていると考えられた.筆者らはS1P2遺伝子座にLacZ遺伝子をノックインしたマウスを樹立・解析し,LacZ活性組織染色によってマウス各組織におけるS1P2発現細胞を同定した.上述のようにS1P2は培養血管内皮細胞にはほとんど発現していないが,多くの臓器で血管平滑筋とともに血管内皮細胞にS1P2が発現していた.腫瘍内では血管細胞の他に,S1P2は浸潤している骨髄系細胞にも発現していた.S1P2-KOマウスから単離した血管内皮細胞では,増殖,細胞遊走,毛細血管様構造形成能のいずれも,野生型内皮細胞に比較して亢進を示した.S1P2-KOマウスの内皮細胞では,細胞運動の分子スイッチである低分子量Gタンパク質Racおよび細胞増殖に関わるリン酸化酵素Akt活性の亢進がみられた.さらに,KOマウス内皮細胞を腫瘍細胞とともに皮下移植したところ,移植内皮細胞は新生腫瘍血管の内皮に組み込まれ,野生型内皮細胞の移植に比較して血管新生および腫瘍増殖はともに亢進した.これらの結果より,内皮細胞のS1P2は,腫瘍増殖,血管新生を抑制すると考えられた.また,S1P2-KOマウス骨髄の移植を受けた野生型マウスでは腫瘍増殖と血管新生が亢進したことから,内皮のS1P2のみならず,骨髄由来細胞のS1P2も腫瘍血管新生および腫瘍増殖に抑制的に働くことが示唆された.
3)S1P2による動脈硬化の促進
高血圧,高脂血症,肥満,喫煙などにより血管内皮が傷害されると,単球/マクロファージやTリンパ球が傷害された内皮細胞に接着し内皮下に浸潤する34).単球はマクロファージに分化し,スカベンジャー受容体を介して酸化LDL(OxLDL)を細胞内に取り込んで泡沫細胞となる.これら炎症性細胞と内皮細胞は,腫瘍壊死因子(TNF-α),インターフェロン-γ(INF-γ)などの炎症性サイトカインや,MCP-1などのケモカインを産生し,血管内皮機能障害の増悪,単球浸潤とマクロファージ泡沫化の促進,および中膜血管平滑筋の内膜への遊走・増殖・細胞外マトリックスの蓄積を引き起こす.この結果,内膜肥厚・内腔狭窄が進展する.
Hlaらは,S1P2が粥状動脈硬化病変の形成に促進的に働いていることを明らかにした35).S1P2-KOマウスをApoE-KOマウスと交配してApoE-KO背景とし,高コレステロール食を負荷した.S1P2-KOマウスでは野生型マウスに比べて大動脈の粥状動脈硬化病変面積が約70%抑制された.高コレステロール血症の程度は,両マウスで差が認められなかった.このKOマウスにおける動脈硬化抑制の程度は,これまでの粥状動脈硬化研究で報告されたさまざまな遺伝子のKOマウスの中でも,最も顕著な効果が観察された例である.S1P2-KOマウスの粥状動脈硬化病巣では,マクロファージ浸潤が減少した.S1P2-KOマウス骨髄を野生型マウスに移植したキメラマウスでは,S1P2+/+マウス骨髄を野生型マウスに移植したコントロールマウスに比較して粥状動脈硬化病変面積が減少し,マクロファージのプラーク内密度も低下した.これらの結果から,骨髄由来細胞,特に単球/マクロファージに発現するS1P2が粥状動脈硬化病変の形成に重要な役割を果たしていることが示唆された.
血管傷害に伴う新生内膜形成に対しては,S1P2が逆に抑制作用を及ぼすことが報告された36).マウス頸動脈の結紮によって引き起こされる内膜肥厚は,S1P2-KOマウスで著しく増強した.S1P2-KOマウスの頸動脈内膜と平滑筋層では,分裂細胞が著明に増加した.S1P2-KOマウスおよび野生型マウスの頸動脈から単離した平滑筋細胞のS1Pに対する化学遊走は,S1P2-KOマウスから採取した平滑筋細胞で促進された.
4)S1P2による血管トーヌス増大
内皮細胞に発現するS1P1,S1P3はNO産生を高めて血管拡張を引き起こす37)のに対し,血管平滑筋に発現しているS1P2,S1P3は血管収縮を引き起こすので,多くの血管床ではS1P投与により血管収縮が観察される.S1P2はこの他内皮にも発現し,上述したようにNO産生を抑制している24, 38).S1P2-KOマウスでは,血圧の基礎値は野生型マウスより低値である28, 39).平滑筋および内皮のS1P2はいずれもその作用機構の上からは血圧に影響を及ぼしうるが,eNOS遺伝子を欠損したマウスでもS1P2 KOは血圧の基礎値を低下させるので,少なくとも血管平滑筋に発現するS1P2は血管トーヌス増加に寄与すると考えられる28).
血圧に大きな影響を及ぼす抵抗血管ではSphK1が血管平滑筋トーヌスに関与することが報告されている.血管平滑筋でSphK1により産生された局所メディエーターとしてのS1Pは細胞内Ca2+濃度の上昇を引き起こすとともに,Rhoを活性化する.RhoはRhoキナーゼを活性化して20 kDa軽鎖ホスファターゼを抑制するので,Ca2+によって活性化されたミオシンリン酸化酵素(ミオシン軽鎖キナーゼ)により引き起こされた20 kDa軽鎖のリン酸化レベルは増強される40).
S1P2-KOマウスは内耳性の聴力障害を呈する41, 42).内耳蝸牛内を灌流するspiral modiolar arteryには,S1P1~S1P3,SphK1が発現し,外因性にS1Pを作用させるとRho, Rhoキナーゼを介して収縮が増強される.聴力障害は,S1P2を欠損するとS1Pに対する平滑筋の収縮不全によりspiral modiolar arteryのトーヌスが低下し蝸牛内血管に過剰な血流が流れて毛細血管が障害され,その結果蝸牛血管条の上皮が障害される結果と考えられる43).
心不全では全身の血管のトーヌスが不適切に増加しており,これは不全心への負荷となって心不全を悪化させる一要因となる.心不全では,血管平滑筋のSphK1およびS1P2を含むS1P受容体がTNF-αと協働して血管トーヌス増加に関与すると報告されている44).このように,炎症などでTNF-α産生が増加している状態では,血管壁におけるSphK1活性化–S1P産生–S1P受容体活性化の経路を介して血管トーヌスが増加することが示唆される.
5)S1P2によるリンパ球の遊走と局在の調節,リンパ腫発症の抑制
S1P2はS1P高濃度を忌避して遠ざかる化学反発(chemorepulsion)を媒介する受容体である.また,Rho-Rhoキナーゼの下流でAktを抑制することから,細胞増殖抑制作用を及ぼす場合がある.二次リンパ組織の胚中心へのBリンパ球の局在化と増殖抑制にS1P2が関与することが示されている45).また,S1P2はヘルパーTリンパ球の局在化にも関与する46).
S1P2-KOマウスはびまん性大細胞型B細胞性リンパ腫(DLBCL)を発症し,上記のBリンパ球に及ぼす作用においてはS1P2は腫瘍抑制遺伝子として機能している47).さらに,ヒトDLBCL症例の一部ではS1P2遺伝子に変異が認められ,この変異はS1P2のリン酸化酵素Aktの活性化抑制および細胞遊走抑制(化学反発)作用を消失させるloss-of-function変異である48).これはS1P2がRho活性化作用を失った結果であり,三量体Gα13あるいはRho活性化GEFであるARHGEF1のノックアウトも類似の表現型を来す.
6)S1P2によるてんかんの抑制
MacLennanら49)は幼若なS1P2-KOマウスが痙攣発作(てんかん)を呈し,離乳までに相当数のマウスが死亡することを最初に報告した.痙攣時および非痙攣時のいずれにおいても脳波異常がみられ,脳スライスのパッチクランプ法によって新皮質錐体神経細胞の過剰な興奮性が観察された.その後,筆者らを含む数グループによって樹立された複数系統のS1P2-KOマウスにおいて,C57BL/6マウスとの戻し交配(バッククロス)の回数を重ねるとてんかん発作が出現することが判明した.Akahoshi, Ishiiら50)は,てんかん発作を詳細に解析した.連続ビデオモニタリングにより,20匹の3~8週齢のS1P2-KOマウス(C57BL/6マウスと7回戻し交配)のうち17匹で,生後25日~45日の間にてんかん発作がみられた.てんかん発作はミオクローヌス発作で開始し,その後に数秒間走り回り動かなくなった.一部のマウスは走り回ったあとに強直間代性の痙攣を呈しその後動かなくなった.マウスは多くの場合にはこの後回復したが,一部のマウスは呼吸停止により死亡した.S1P2-KOマウスの非発作時の脳波記録では,同期した高電位棘波が広範囲に観察された.多数のS1P2-KOマウスの観察から,生後3.5~6週の間に224匹中90匹(40.2%)と高率に死亡したことがわかった.S1P2 mRNAの発現は海馬の錐体神経細胞と顆粒神経細胞に認められ,脳内の他の部位では検出されなかった.
7)S1P2の線維症への関与
肺,肝臓や心筋の線維化(臓器線維症)は,慢性炎症に伴う線維芽細胞の増生と線維芽細胞によるコラーゲン線維の沈着が生じ,線維沈着の進行により正常な組織構造が破壊されて機能障害を来す病態である.臓器線維症には,マクロファージをはじめとする炎症細胞や線維芽細胞の活性化が関与すると考えられているが,いまだ不明の点が多い.
胆管結紮により胆汁流出を阻害すると肝臓の線維化を来す.S1P2選択的アンタゴニストであるJTE-013の慢性投与あるいはS1P2-KOにより,胆管結紮による肝臓線維化が抑制された51).S1P2遺伝子座にLacZをノックインしたマウスを用いて肝臓におけるS1P2の発現細胞を検討したところ,S1P2は動静脈の平滑筋の他に,類洞周辺の肝星細胞に発現していた.肝星細胞は類洞血管の周皮細胞に相当する細胞であり,肝線維化に際して活発に遊走・増殖して筋線維芽細胞の形質を呈することが知られている.線維化を来した肝では,線維化部位の平滑筋特異的αアクチン(α-smooth muscle actin:αSMA)陽性の筋線維芽細胞にS1P2の高発現を認めた.これらの結果から,胆管結紮肝線維化モデルでは,肝星細胞のS1P2が筋線維芽細胞への転換に関与することによって線維化を促進する可能性が示唆された.四塩化炭素(CCl4)投与による肝障害後線維化モデルにおいてもS1P2-KOおよびS1P2アンタゴニストは線維化を抑制した52).
ブレオマイシン投与による肺線維症がS1P2-KOマウスで抑制されることを筆者らは見いだした53).ブレオマイシン投与は線維化が一過性となる気道内1回投与ではなく,線維化が持続する腹腔内反復投与(2回/週,3週間)のプロトコールを用いた.LacZノックインマウスにおいて,S1P2発現は線維化肺の肺胞マクロファージ,肺胞上皮,肺血管内皮などに認められた.骨髄移植実験により,S1P2を欠損する骨髄を移植されたマウスでは肺線維化が軽減した.また,マクロファージを枯渇させると肺線維化が軽減したことから,マクロファージは線維化発症に密接に関わることが示された.ブレオマイシン投与は,肺胞マクロファージにおけるIL-13, IL-4といった線維化誘発作用のあるサイトカインの発現や2型マクロファージマーカーであるArginase1, Fizz1, CCL17などの遺伝子発現を促進し,S1P2-KOはこれらの遺伝子発現を著しく抑制した.ブレオマイシン反復投与肺線維症モデルでは,IL-13作用を阻害すると肺線維化は強く抑制された.ブレオマイシン投与マウスから採取したマクロファージでは,IL-13によりリン酸化・活性化される転写因子STAT6のリン酸化が増加していたが,S1P2-KOマウスから採取したマクロファージではSTAT6リン酸化がかなり抑制された.IL-13受容体分子のIL13Rα1, IL4Rαの遺伝子発現には変化がなかったので,S1P2-KOマウスでのSTAT6リン酸化抑制はIL-13受容体活性化以降のメカニズムによると考えられた.S1P2選択的アンタゴニストの慢性投与により,肺線維化は抑制された.