2. Non-EDG型LPA受容体に対するリガンドの特徴
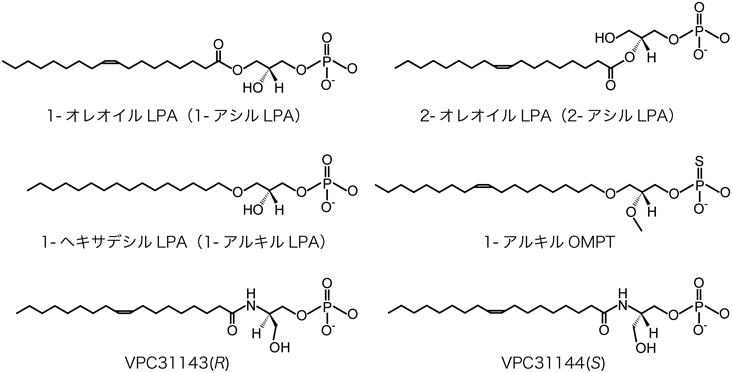
LPAは単一の化合物を指す言葉ではなく,炭化水素鎖の化学的性質およびグリセロール骨格への結合が異なる多くの分子種の総称である15).各LPA受容体は,さまざまなLPA分子種に対して異なるリガンド選択性を有するのが特徴である.たとえば,LPA3は,グリセロールのsn-1位置(1-アシルLPA)と比較して,sn-2位(2-アシルLPA)にアシル鎖を有するLPAによってより強力に活性化される16).この節では,Non-EDG型LPA受容体のリガンド選択性について議論し,いくつかのLPA類縁体(アナログ)や非脂質様構造を持つ化合物のアゴニスト/アンタゴニスト効果についても言及する.
1)LPA4のリガンド選択性
いくつかのLPA分子種に対するLPA4の反応性は,LPA4を発現させた細胞を使ったレポーター遺伝子実験で調べられている7).1-アシルLPAの受容体活性化強度は,1-オレオイル(18 : 1)>1-ステアロイル(18 : 0)>1-パルミトイル(16 : 0)>1-ミリストイル(14 : 0)の順であった.飽和脂肪酸よりも不飽和脂肪酸を有するLPAの方が,また炭素鎖の長い脂肪酸の方が活性は強いようである.さらに,1-アシルLPAの方が1-アルキルLPAや1-アルケニルLPAよりもLPA4活性強度は高かった.LPA4を発現させた細胞の膜画分を用いた放射性リガンド結合実験でも同様の結果が得られている.
2)アルキルLPAへの選択性を示すLPA5
LPAはヒト血小板の形状変化を直接誘導する17, 18).より高濃度のLPA存在下では血小板からADPの放出が促され,LPAとADPは協調して血小板凝集を引き起こす.さまざまなLPA分子種に対するヒト血小板の応答(形状変化およびその後の凝集)の強弱を調べた以前の研究により,血小板のLPA反応特異性はEDG型LPA受容体を介した応答によっては形成されないことが示唆されていた.すなわち,EDG型LPA受容体が示すLPA分子種の構造活性相関16)とは対照的に,血小板は1-アシルLPAよりも1-アルキルLPAに高い反応性を示すのである18–20).LPA4とLPA5のmRNAはヒト血小板に豊富に発現しているため21, 22),これら二つのNon-EDG型LPA受容体のいずれかまたは両方がLPAによる血小板活性化に関与している可能性があるが,近年の研究からLPA5が血小板のLPA分子種への反応特異性を規定することが明らかになってきた.血小板形状変化反応とLPA5を外来性に発現した培養細胞のカルシウム流入反応について,複数のLPA分子種で反応を惹起したところ両反応で活性化強度の順序が一致している[1-O-オレイル(18 : 1)≥1-O-ヘキサデシル(16 : 0)>1-オレオイル(18 : 1)≫1-O-オクタデシル(18 : 0)]21).さらに,LPA5のアゴニスト21, 23)およびアンタゴニスト活性23, 24)を示すさまざまな化合物を用いた実験においては,LPA5活性化とヒト血小板形状変化は一貫して関連している.siRNAを用いた実験でも,ヒトDami巨核芽球細胞およびMeg-01巨核芽球細胞のLPA誘発性の形状変化がLPA5によってもたらされることが示されている25).LPAは,酸化LDLおよびヒト動脈硬化性病変の主要な血小板活性化脂質であると同定されていることから26),LPA5は抗血栓療法の新規標的分子となる可能性がある.なおLPA5の機能と関連があるか不明であるが,アルキルLPAと反応させても凝集しない血小板を持つヒトがある割合で存在するとの報告がある20, 27).この「LPA非応答者」は,冠状動脈疾患に罹患する割合が有意に低いことも併せて報告されている27).この点でもLPAによる血小板凝集は臨床的に興味深い.
3)2-アシルLPAへの選択性を示すLPA6
筆者らは,LPA6を発現させた培養細胞でリガンド選択性を調べた10).1-アシルLPA分子種では,1-リノレオイル(18 : 2)>1-オレオイル(18 : 1)>1-アラキドノイル(20 : 4)=1-パルミトイル(16 : 0)≫1-ステアロイル(18 : 0)=1-ミリストイル(14 : 0)の順にLPA6活性化能が高く,不飽和脂肪酸を有するLPAの方が飽和脂肪酸を有するものよりも強いアゴニストであると考えられた.さらに,不飽和脂肪酸を有するLPA分子種の中で,2-アシルLPAは1-アシルLPAよりも強いアゴニストであった.
膜結合型ホスファチジン酸選択的ホスホリパーゼA1α(mPA-PLA1α)は2-アシルLPAを産生する酵素である28).mPA-PLA1αおよびLPA6のmRNAはともに,ヒト毛根の内毛根鞘において発現する29, 30).よって,mPA-PLA1αによって産生される2-アシルLPAは毛包内のLPA6の内在性リガンドとして作用する可能性がある.毛髪成長とLPA6の関係については,5-11)項でさらに議論する.
4)LPA受容体に対するLPA以外のリガンド
N-アシルエタノールアミドリン酸(NAEPA)はLPAアナログであり,LPA1とLPA2のアゴニスト作用を持つ31).2種類のNAEPAの誘導体VPC31143(R)およびVPC31144(S)は鏡像異性体である(図1)32).筆者らは,培養細胞に外来性に発現させた6種類のLPA受容体をVPC31143(R)とVPC31144(S)と反応させて,以下の知見を得た13).1)3種類のEDG受容体とLPA6に対しては,VPC31143(R)の方がVPC31144(S)よりも強いアゴニスト活性を示す.2)これとは対照的にLPA4とLPA5に対しては,VPC31144(S)の方がVPC31143(R)よりも強いアゴニスト活性を示す.上記の立体選択性を利用すれば,LPA4やLPA5によって惹起される生体反応を,それら以外のLPA受容体によるものと区別して観察できるかもしれない.さらに,2-2)項で言及したLPA5に選択性の高い1-アルキルLPAや次の段落で言及するアルキルOMPTを併用すれば,LPA4とLPA5の反応も区別できる可能性もある.
1-オレオイル-2-O-メチル-rac-グリセロチオリン酸(1-アシルOMPT)は,リン酸基の代わりにチオリン酸基を含むLPAアナログである33, 34).LPA3に対する選択的アゴニスト活性を有するとして,1-アシルOMPTとそのsn-1位のアシル基をアルキル基に置換した1-アルキルOMPT(図1)35)は多くの研究で使用されてきた36–38).筆者らは1-アルキルOMPTがLPA6のアゴニストでもあることを見いだした10).その後,Jianらも同様の結果を報告している39).併せて彼らは,1-アシルOMPT, 2-アシルOMPT, 2-アルキルOMPTもLPA3とLPA6のアゴニストとして機能すること,そして1-アルキルOMPTと2-アルキルOMPTが強力なLPA5アゴニストであることを示した.よって,アシル/アルキルOMPTを用いた実験結果の解釈には注意する必要があることが明らかになってきた.また,LPA5がアルキル基を持ったLPAアナログに高い反応性を示す結果は,2-2)項で論じたように,LPA5が「血小板型」LPA受容体であるという考えを支持している.アルキルOMPTはLPAと異なり,ホスファターゼおよびホスホリパーゼの両方による分解から保護されていることから,生体内でのLPA5やLPA6の機能を解明するための研究で有用と考えられる.
ファルネシルモノリン酸(FMP)およびファルネシルピロリン酸(FPP)はLPA3に対する内在性アンタゴニストとして報告されていた化合物である40).その後,両化合物はLPA4に対してはアンタゴニスト活性を,LPA5に対してはアゴニスト活性を持つことが明らかにされている23, 41).FPPはLPA6を活性化するという報告もある42).
Ki16425は非脂質様構造を有し,三つすべてのEDG型LPA受容体に拮抗作用を及ぼす化合物である43).ただし,LPA2への拮抗作用は弱い.Ki16425およびその構造アナログKi1619844)は,Non-EDG型LPA受容体のどれにもアンタゴニスト活性を示さない8, 10, 44, 45).したがって,二つのLPA受容体ファミリーの間でLPAの認識様式は異なることが予想される.AM966も非脂質様構造を有する経口投与可能なLPA1受容体アンタゴニストである46).この化合物は,LPA4およびLPA5を含む他のLPA受容体よりもLPA1受容体に対して選択性がかなり高い.LPA6に対するアンタゴニスト活性については不明である.
3. Non-EDG型LPA受容体のGタンパク質共役の選択性
EDG型LPA受容体のGタンパク質共役については多くの研究がある.LPA1とLPA2はGi,GqおよびG12/13タンパク質と共役し,LPA3はGiおよびGqタンパク質と共役する4).以下,Non-EDG型LPA受容体のGタンパク質共役選択性に関して議論する.表1には概略を示す.
表1 Non-EDG型LPA受容体のGタンパク質共役(文献13より引用,一部改変)| 受容体サブタイプ | 共役するGタンパク質 | 内在性の受容体との共役が報告されている細胞 |
|---|
| LPA4 | G12/13 (+) | マウス胎仔繊維芽細胞48) |
| Gs (+) | HT1080線維肉腫細胞52),ヒト間葉系幹細胞53) |
| Gq (+) | なし |
| Gi (+) | なし |
| LPA5 | cAMP↑(+)* | B16メラノーマ細胞58) |
| Gq (+) | ラット初代培養後根神経節ニューロン41),LAD2マスト細胞60) |
| G12/13 (+) | Dami巨核芽球細胞25),Meg-01巨核芽球細胞25) |
| LPA6 | G12/13 (+) | ヒト臍帯静脈血管内皮細胞10),マウス初代培養ケラチノサイト61),HaCaTケラチノサイト61),HEK293細胞61) |
| Gi (±) | なし |
| Gs (±) | なし |
(+):複数の独立した研究により支持されている結果.
(±):さらなる検証が必要と思われる結果.
*非古典的な,Gs非依存性経路によるものと思われる結果. |
1)LPA4のGタンパク質共役
LPA4を外来性に発現させた培養細胞において,LPA4がG12/13タンパク質を介してRhoシグナル伝達経路を活性化することが複数の独立した研究で示されている45, 47).LPAによる細胞形態変化はG12/13のミニ遺伝子,Rho阻害剤(C3 exoenzyme)またはRhoキナーゼ(ROCK)阻害剤(Y27632)に感受性であった.LPAによるRhoの活性化は,LPA4を内在的に発現するマウス胚線維芽細胞(MEF)でも観察されるが,LPA4-ノックアウト(KO)胚から単離したMEFではこの活性化が著しく減弱していた48).後に5-1)項で議論するように,一部のLPA4-KO胚は血管形成不全を示し49),G13-KO胚やG12/G13二重ヘテロ胚と同様の表現型であった50, 51).以上の結果は,LPA4の重要なシグナル伝達がG12/13タンパク質とRhoに依存的であることを強く示唆する.
外来性に発現させたLPA4ではG12/13タンパク質との共役に加えて,サイクリックAMP(cAMP)産生の観点からGsタンパク質にも共役すると考えられる論文が報告されている7, 45).その論文のうち,LeeらはGsミニ遺伝子を細胞に導入してLPA4によるcAMP産生にGsタンパク質が関与することを示した45).内在性に発現するLPA4についても,Gsタンパク質との共役が示唆されている.たとえば,ヒトHT1080線維肉腫細胞においてLPAがLPA4依存性に浸潤突起を誘導することを示した論文では,がん細胞が浸潤する際に形成されるこの膜構造が細胞内cAMPの増加を介して作られることから,LPA4がcAMP産生に関わることを提唱している52).また別の論文では,骨芽細胞へ分化中のヒト間葉系幹細胞がLPA1とともにLPA4を発現することを示した上で,LPAがLPA1非依存的に細胞内cAMPレベルを増加させることを示している53).この細胞でLPA4の発現をノックダウンすると,細胞の骨形成分化を増強することは注目に値する.一方で筆者らの実験では,生来LPA4を高発現するMEFをLPAで刺激しても,cAMP産生応答を観察することはできなかった(未発表データ).さらに,細胞株にLPA4を発現させた実験では,チャイニーズハムスター卵巣(CHO)細胞ではLPAはcAMP産生を認めたが7),B103細胞では認められなかった47).LPA4を介したcAMP産生に関して一貫性が乏しいのは,Gsタンパク質とLPA4の共役効率や活性化するアデニル酸シクラーゼのアイソフォームの種類が,細胞の種類や培養条件,LPA4の発現レベルなどによって左右されるのが一因かもしれない.実際,リガンド刺激後に起こる受容体のリン酸化によって,共役するGタンパク質の種類がGsからGiにスイッチする現象が明らかになっている54).また,アデニル酸シクラーゼにはGsタンパク質による過敏化/不応化の様式が異なる複数のアイソフォームが存在する55).
LPA4がGiおよびGqタンパク質と共役することは,やはりLPA4を培養細胞に発現させた研究結果に基づく7, 45, 47).GiおよびGqタンパク質との共役は,それぞれ百日咳毒素(Giタンパク質阻害剤)とYM-254890(Gqタンパク質阻害剤)またはGqミニ遺伝子で処理したときの影響を観察することで確認している.LPA刺激後の細胞応答(Giタンパク質ではcAMP産生抑制,GiおよびGqタンパク質では細胞内カルシウム流入反応)が減弱するとき,それぞれのGタンパク質と共役すると判断されている.一方,内在性LPA4のGiおよびGqタンパク質との共役については報告がない.たとえば,野生型およびLPA4-KO胚に由来するMEFにおいて,筆者らはLPA誘導性のアデニル酸シクラーゼ阻害およびカルシウム流入反応の差異は観察できていない(未発表データ).生理学的状況における内在性LPA4のGiおよびGqタンパク質への共役は依然として不明確であり,さらなる検証が必要であろう.
2)LPA5のGタンパク質共役
外来性または内在性に発現するLPA5は,いくつかの細胞株においてLPA誘導性の細胞内cAMP産生を媒介することが示されている8, 23, 41, 56, 57, 58).しかしながら,二つの独立した研究によって,LPA5によるcAMP産生がGs以外のGタンパク質と共役することにより起こる可能性が示されている.Leeらは,LPA5を発現させた培養細胞において,LPAによるcAMP産生はGsミニ遺伝子によって阻害されないことを報告した8).Jongsmaらは,LPA5を内在的に発現するマウスB16メラノーマ細胞において,蛍光共鳴エネルギー移動に基づくcAMPセンサーを用いて,LPA5依存性の細胞内cAMP増加を詳細に分析した58).この細胞におけるLPA5刺激後の細胞内cAMPの増加は一過性であり,Gs依存性に細胞内cAMPを持続的に増加させることが知られるα-メラニン細胞刺激ホルモンへの応答とは明らかに異なっていた.
外来性に発現させたLPA5とGqタンパク質との共役(Gqミニ遺伝子感受性または百日咳毒素耐性の細胞内カルシウム流入反応やホスファチジルイノシトール分解反応)は,いくつかの細胞株で観察されている8, 56, 59).内在性LPA5のノックダウン実験は,ラット初代培養後根神経節ニューロン41)およびヒトLAD2マスト細胞60)におけるLPA誘発カルシウム流入反応におけるLPA5の関与を示した.
LPA5を外来性に発現する細胞では,LPAによる細胞形態変化がG12/13とROCK依存性であることが観察されている8).さらに,二つのヒト巨核芽球細胞(Dami細胞およびMeg-01細胞)におけるROCK依存性のLPA誘発形態変化は,内在性LPA5のノックダウンによって阻止された25).また,ヒト血小板に対してLPAは,Rho–ROCK経路を活性化して形状変化をもたらす17, 18).血小板のLPA応答にもLPA5とG12/13タンパク質が関わっているものと思われる.
3)LPA6のGタンパク質共役
筆者らを含む三つの独立したグループによる研究により,LPA6がG12/13–Rho–ROCK経路を活性化するというコンセンサスが確立されている10, 42, 61).筆者らは,LPA6を外来性に発現する細胞において,LPA6がROCK依存的に細胞骨格変化を誘導することを見いだした10).LPA6がG13と共役することは,G13共役受容体によるアデニル酸シクラーゼ活性化を可能にする「Gαs/13キメラタンパク質」を用いて示した.LPA処理後の細胞膜画分から免疫沈降させたGα13タンパク質が,[35S]GTPγS標識されることも確認した.内在性LPA6がRhoの活性化に関わることは,LPAによって誘導されるヒト臍帯静脈血管内皮細胞(HUVEC)の細胞形態変化がLPA6 siRNAによって抑制されることで示した.Leeらによる独立した実験でも,LPA6とSREレポーター遺伝子を外来性に発現させた細胞において,LPA6がG12/13–Rho–ROCK経路を活性化していた42).彼らの実験では,LPAによるレポーター遺伝子の発現上昇はG12/13阻害剤(p115RhoGEFのGタンパク質シグナル伝達調節ドメイン),C3 exoenzymeまたはY27632のいずれによっても阻害された.また,Inoueらによる研究ではLPA6を外来性に発現する細胞において,膜結合型TGF-α前駆体のエクトドメインをLPAがROCK依存的に切断することを観測した61).また,G12とG13タンパク質の二重KO細胞では,LPA6によるTGF-αの切断はほぼ抑制された62).また,ヒトHaCaTケラチノサイトの内在性LPA6のノックダウンでもLPAによるTGF-αの切断は抑制された61).
Leeらは,LPA6を発現させたCHO細胞において,フォルスコリンによって誘発された細胞内cAMPレベルの上昇をLPAが阻害することを報告した42).しかし,筆者らはLPA6を発現させた別の細胞(B103細胞とRH7777細胞)において同様の応答を誘導することはできていない10).ただし,LPA6を発現させたB103細胞におけるGαs/13キメラタンパク質依存性のcAMP増加は,細胞を百日咳毒素で前処理した場合にのみ観察された.この結果は,LPA6がGiを含むいくつかのGタンパク質と共役し,Giを介する応答がアウトプットの応答(キメラGタンパク質を介したアデニル酸シクラーゼの活性化に起因するcAMP産生)をキャンセルしていたことを示唆している10).一方,Gsタンパク質との共役についてはLPA6を発現させたCHO細胞でLPA6が細胞内cAMPレベルを増加させた報告があるが9),他の独立した研究によっては追試されていない.GiおよびGsタンパク質に対するLPA6の共役に関するコンセンサスを確立するためには,さらなる検証が必要であろう.
KOマウスの研究から,現在までにさまざまな臓器および発生段階におけるNon-EDG受容体の生理学的役割が明らかになってきた.in vitroおよびin vivoの実験を通して分子機構の解明も進んでいる.
1)血管新生とLPA4
2006年に二つの研究グループより,ATX-KO胚が重度の血管形成不全を伴って死亡する論文が発表された64, 65).これらの表現型はその後,別の研究グループによっても追認されている66, 67).触媒的に不活性なATXノックインマウスは,ATX-KOマウスと同様の表現型を示す68).この結果は血管形成不全の表現型がATX依存性LPA産生の喪失に起因することを示している.G13-KOマウスやG12/G13二重ヘテロマウスが同様に血管形成不全で胎生致死に至る結果50, 51)は,血管形成に必須なG12/13共役型のLPA受容体の存在を示唆している.加えて,血管内皮細胞(EC)特異的G13-KO胚でも同様の致死性の血管形成不全を呈することを考慮すると69),このLPA受容体はECで機能する可能性が高い.なお,EDG型LPA受容体の中でLPA1およびLPA2はG12/13タンパク質を活性化することが知られているが4),LPA1,LPA2,LPA3の単一および重複KOマウスで血管形成不全を表現型として言及した報告はない70–73).したがって,「血管形成に寄与するのはNon-EDG型LPA受容体かもしれない」という仮説が成り立つ.実際,一部のLPA4-KO胚(約30%)は胎生致死であった49).興味深いことに,LPA4-KO胚ではいくつかの臓器で出血が認められ,血管の組織学的な観察では異常な拡張に加えて平滑筋細胞および周皮細胞の動員不全も認められた.LPA4 mRNAは血管のin situハイブリダイゼーションによりECに発現することが示されている.したがって,ECに発現するLPA4はおそらく正常な血管の発達に重要であり,血管新生におけるATXとG12/13の機能を結びつける分子である可能性がある.ただし,LPA4-KOマウスで観察された致死の表現型は浸透率が約30%と低く,ATX-KOマウスで100%の胚が致死であるのと対照的なことは慎重に考慮すべき点である.これに関しては,別のG12/13共役LPA受容体がLPA4と協調して血管新生に関与することで説明がつくかもしれない.
筆者らが樹立したLPA4-KO胚の中には,全身に浮腫を呈するものがあった49).これらの胚ではリンパ管が拡張していたことから,LPA4は血管形成以外にもリンパ管形成において重要な役割を持つことが考えられた.ATX-KO胚のリンパ管の欠損は報告されていない.これはATX-KO胚が,マウスのリンパ管形成時期(胎生11.5日前後)74)にはすでに死亡しているためであると考えられる.
2)高内皮小静脈(HEV)とLPA4
リンパ節やパイエル板などの二次リンパ組織内には高内皮小静脈(HEV)と呼ばれる特殊な静脈が構築されており,血中のナイーブリンパ球はHEVの内皮細胞層を通り抜けて二次リンパ組織の実質へと移動する75).HEVのECに発現するLPA4がリンパ球血管外遊出に促進的に働くことが論文発表されている76).LPA4-KOマウスのHEVでは,EC層とその下の基底膜の間に蓄積されるリンパ球(二次リンパ組織の実質へ移動する直前のリンパ球)の数が上昇していた.この表現型が現れる機序は未解明であるが,ECのLPA4シグナルが消失するとリンパ球との相互作用が増強してしまうか,もしくはECの運動性が悪くなってしまうためにリンパ球の移動が妨げられる可能性をこの論文では考察している.
3)腫瘍血管とLPA4
マウスにがん細胞を移植して形成させた固形腫瘍の毛細血管では,蛇行や無秩序な分岐などの異常が認められ,血流も少ない.この担がんマウスにLPAを投与すると,異常な血管網が正常組織と同様のものに変化することが報告された77).LPAの有意な効果は短時間のうち(24時間以内)に観測されることから,ECの増殖を促すというよりも形態変化(伸長)を促すことがLPAの具体的な働きと考えられる.LPAは腫瘍の毛細血管の血流を増やし,がん組織へ運搬される血液成分も増やしていた.腫瘍中のECでは,6種類のLPA受容体のうちLPA4とLPA6のmRNAが主に発現していた.そこで,LPA4-KOマウスを担がんマウスに用いたところ,上述のLPAの腫瘍血管への作用は消失した.一方で,LPA6-KOマウスは野生型マウスと同様のLPAへの反応性を示した.これらの結果から,ECにおけるLPA4シグナルが腫瘍血管の機能の正常化に寄与していることが示唆された.マウスに移植したがん細胞の増殖を抗がん剤が抑える効果をLPAは増強したことから,LPA4活性化に伴う腫瘍血管の正常化は抗がん剤治療に資する可能性がある.マウス膵島EC由来のMS-1細胞を使ったin vitro実験では,LPA4はGiとG13を介してVE-カドヘリンの細胞膜移行と細胞膜直下でのストレスファイバー形成を促し,この現象がEC間のバリア機能を亢進させる可能性を示した.ただし,VE-カドヘリンの細胞膜移行に至る細胞内シグナル経路は明らかになっていない.一方,Renらはヒト肺動脈血管内皮由来のEC(HPAEC)をLPA刺激した際に,EC間のバリア機能がLPA6依存的に低下することを報告している(5-10)項でも言及)78).腫瘍血管のECやMS-1細胞におけるLPA4の(LPA6に対する相対的)発現レベルはHPAECより高いようであり77, 78),このことが同じECに分類される細胞でLPAに対する応答性が異なった原因になるのかもしれない.また,LPA4とLPA6を含めLPA受容体6種類の発現プロファイルの違いによって,LPAに対するECの応答性が変わった可能性もある.
4)造血とLPA4
骨髄は哺乳動物の主要な造血器官である.すべての成熟血球細胞は,骨髄の造血幹/造血前駆細胞から作られる.造血幹/造血前駆細胞の維持,分化,および増殖には,周囲の細胞が作るサイトカインをはじめとした分子群の働きを必要とする.これら分子が形成する局所的な微小環境は,骨髄ニッチと呼ばれる79).骨髄に存在する間葉系の間質細胞はニッチの構築に重要な役割を持つ.筆者らは,マウス骨髄におけるLPA4 mRNA発現が,造血幹/造血前駆細胞よりも血小板由来増殖因子受容体α(PDGFRα)陽性の間質細胞でかなり高いことを観察した80).マウスに致死量に近い量の放射線照射や抗がん剤投与を施すと,造血幹/造血前駆細胞の分化/増殖の障害(骨髄抑制)によって末梢血液の成熟血球細胞数が減って死亡する個体が出るが,LPA4-KOマウスは野生型マウスに比べて末梢血液中の白血球数が早く減り,致死率も高かった.また,骨髄抑制で一度減少した骨髄の造血幹/造血前駆細胞数の回復が,LPA4-KOマウスでは遅延していることもわかった.野生型マウスの骨髄に置換したLPA4-KOマウスでも,骨髄抑制による致死率の高さが再現できた.よって,骨髄間質細胞が発現するLPA4が骨髄抑制後の造血回復,すなわち造血幹/造血前駆細胞の維持・分化・増殖に寄与する骨髄ニッチ関連LPA受容体であることが示唆された.骨髄間質細胞で発現するLPA4が,造血幹/造血前駆細胞の増殖に関わる幹細胞因子(SCF)81)の生産を調節する可能性をin vitroとin vivoの実験で示したが,この調節に関わる細胞内シグナルについては未検討である.
5)骨形成とLPA4
LPA4 mRNAは骨において高いレベルで発現している53).LPA4-KOマウスでは野生型マウスと比較して骨密度が増加することを報告したLiuらは,LPA4がGsタンパク質の活性化および細胞内cAMPレベルの増加を介して,骨芽細胞の分化を阻害すると考察している53).一方,LPA1-KOマウスは骨形成低下の表現型を示し82),LPA4-KOマウスの表現型とは対照的であった.LPA1-KOマウスには,異常な顔面骨の発達も観察されている70).したがって,骨形成ではLPA1とLPA4のシグナルは拮抗する可能性がある.間葉系幹細胞や骨芽細胞でLPA1がGiタンパク質と共役しLPA4がGsタンパク質と共役するならば,LPA1-KOマウスとLPA4-KOマウスで相反する骨の表現型は,細胞内cAMPレベルの相反する調節によって説明できる.in vitroの研究でも,LPA1とLPA4を内在性に発現するMEF48)と,LPA1またはLPA4を外来性に発現させた培養細胞47)において,両受容体の相反する機能が観察されている.これらの研究ではLPA1およびLPA4が,それぞれGi–Rac経路およびG12/13–Rho経路を活性化することで,細胞骨格形態変化および走化性において相反した役割を担っていた.この機序によっても,LPA1-KOマウスとLPA4-KOマウスに現れた相反する骨の表現型を説明できるかもしれない.
6)マスト細胞活性化とLPA5
LPAがヒト初代培養マスト細胞の分化・増殖や活性化に寄与することは,複数の研究で報告されてきた36, 83).LPAはROCK依存的にラット腹腔マスト細胞からのヒスタミン遊離を誘導し84),マウスにおいては痒みによる掻破行動を引き起こす85).LundequistとBoyceは,ヒトLAD2マスト細胞とヒト初代培養マスト細胞において,LPA5のmRNAとタンパク質が豊富に発現することを報告した60).LPAは,LAD2細胞において細胞内カルシウム流入反応およびMIP-1βの放出を誘導するが,これらの応答はLPA5のsiRNAノックダウン後に消滅した.この結果は,LPA5がヒトマスト細胞におけるLPA応答を媒介する主要なLPA受容体サブタイプであることを強く示唆している.ヒトHMC-1マスト細胞におけるLPAよるMCP-1放出反応も,LPA5アンタゴニストで抑制されることが示されている86).ATXはヒト腸管マスト細胞に豊富に発現し87),LPA5 mRNAの発現レベルも腸管で高いことが報告されている8, 41, 56, 59).マスト細胞は,アレルギー性腸炎や炎症性腸疾患などの消化器疾患において重要な役割を果たすと考えられているため,LPA5シグナル伝達はATX依存性LPA産生を介してこれらの疾患に寄与する可能性がある.
7)神経因性疼痛とLPA5
痛みは本来,末梢神経→脊髄→大脳へと伝達された際に知覚される侵害受容性疼痛を指す.しかし,この体性感覚伝達経路に損傷を受けると上位中枢への体性感覚入力が減弱・消失するのに,自発的な痛みや痛覚過敏,アロディニアが生じることがある.このような病的な痛みを神経因性疼痛と呼ぶ.髄腔に存在するLPAは神経因性疼痛の惹起に関与しており88),この過程におけるATX89),LPA190),LPA391)の役割はKOマウスを用いた研究で明らかにされている.末梢神経損傷後,脊髄から放出されたリゾホスファチジルコリンは,ATXによって細胞外でLPAに変換される.LPAはシュワン細胞においてLPA1を介してG12/13–RhoシグナルとGq/11–PLCβシグナルを活性化し,後根神経脱髄および神経因性疼痛に寄与する.また,メカニズムは不明であるが,脊髄後角や後根におけるLPA3刺激は,ATX依存性にLPA産生を増強することによって神経因性疼痛に寄与することも報告されている.LPA5も後根神経節に高いレベルで発現するが8, 41),LPA5-KOマウスの研究からこのLPA受容体サブタイプも神経因性疼痛の発症に関与していることが明らかになった92).LPA1-KOマウスと同様に,坐骨神経部分損傷によって誘導された神経因性疼痛からLPA5-KOマウスが保護されていたのである.興味深いことに,坐骨神経部分損傷に起因する脱髄は,野生型マウスと同程度にLPA5-KOマウスで生じており,この表現型はLPA1-KOマウスとは対照的であった.ゆえに,神経因性疼痛におけるLPA1シグナルの役割は,LPA5シグナルのものとは異なっていると考えられる.LPA5-KOマウスでは脊髄後角ニューロンのリン酸化CREBレベルが減少していたことから,LPA5に媒介されるcAMP–CREBシグナル伝達が後角におけるシナプス活性を増強して,中枢感作に至ることが示唆されている.
神経因性疼痛の惹起における活性化ミクログリアの関与にも注目が集まっている93).LPAによってマウス初代培養ミクログリアは活性化され,LPA5-プロテインキナーゼD(PKD)シグナルを介して炎症を促進する論文が最近発表された94).この論文の中でLPA5がPKDを活性化するシグナル経路については言及していないが,過去にLPAがG12/13–RhoA–PLCεシグナルを介してアストロサイトのPKDを活性化したとする論文がある95).ミクログリアでも同様にPKDの活性化に対してPLCεの関与があるかもしれない.
新規に開発されたLPA5アンタゴニスト(AS2717638)は,マウス髄腔内へのLPA投与またはラット慢性絞扼損傷による神経因性疼痛モデルに対して,経口投与で鎮痛効果を示す57).AS2717638のLPA5に対するIC50値は0.038 µMと,以前に報告されたLPA5アンタゴニストTCLPA5のIC50値0.8 µM24)よりも20倍以上小さい.TCLPA5とAS2717638はともにLPA1,LPA2,LPA3に対するアンタゴニスト作用がほとんどないことは調べられているが,LPA4とLPA6に対する効果は示されていない.
8)腸上皮による水分吸収とLPA5
マウス小腸上皮細胞におけるNa+/H+交換輸送体3(NHE3)を介したNa+依存性の水分吸収を,LPAは促進させる作用がある96).LPA5にはNa+/H+交換輸送体制御因子2(NHERF2)との結合を介して,NHE3を微絨毛の根元部分の細胞質(端網層)から頂端膜へ移行させる機能がある.したがって,LPAの水分吸収促進には腸上皮細胞のLPA5シグナルが重要であると考えられる.実際,腸上皮特異的LPA5-KOマウスでは,LPAによる水分吸収促進作用は消失している97).コレラ毒素を投与されたマウスの小腸における水分吸収の低下をLPAは有意に回復させることから,LPA5の活性化は重篤な下痢の治療に役立つと期待される.
LPAによるNHE3の微絨毛頂端膜への移行には,NHE3のリン酸化が必要である.LPA5とNHE3を外来性に発現するCaco-2bbeヒト大腸腺がん細胞を用いた実験で,頂端膜に存在するLPA5は上皮成長因子受容体(EGFR)のトランス活性化を引き起こすことが観察された98, 99).またEGFRの下流では,RhoA–Pyk2–PDK1経路およびMEK–ERK経路が独立して活性化され,p90リボソームS6K2によるNHE3のS663のリン酸化に至ることが示されている.
9)リンパ球の抗原受容体とLPA5
B細胞とT細胞は抗原を認識するための受容体として,細胞表面にそれぞれB細胞受容体とT細胞受容体を発現する.LPA5シグナルは,B細胞受容体シグナルを抑制することにより,B細胞の活性化と抗体産生を阻害することが報告されている100).この報告では,LPA5–G12/13–ARHGEF1(p115RhoGEF)シグナル経路がIP3受容体の機能を減弱させて,B細胞受容体刺激時の小胞体からのカルシウム放出反応を低下させることを示している.しかし,IP3受容体の機能が減弱する理由は明らかになっていない.CD8+ T細胞でも,LPA5はT細胞受容体からのシグナルを抑制することが同じ研究グループから報告されている101).LPA5はCD8+ T細胞の増殖やCD25(T細胞の活性化マーカー)の発現にも抑制作用を示す.実際,LPA5の抑制作用が解除されているLPA5-KO CD8+ T細胞を養子移入されたマウスでは,腫瘍免疫が増強していた.これらの結果から,がん細胞が産生するLPAがLPA5を介して適応免疫系の細胞傷害性を減弱させる機構の存在が示唆される.上述したB細胞受容体からのシグナルを抑制する機序と同様の機序で,LPA5はT細胞受容体シグナルを抑制していると思われるが,詳細は不明である.リンパ球の抗原受容体のシグナルを抑制するシステムはすでに多く知られているが,LPA5もそのシステムの一つとして自己免疫を防ぐなど正常な免疫系の構築に寄与すると考えられる.
10)血管透過性亢進とLPA6
LPAはストレスファイバー形成によってEC間の接着を緩め,これにより血管の透過性を亢進させて血液成分の漏出を増やす作用がin vitroで報告されている102).HUVECに対してLPAは単層の細胞シートの透過性を亢進させるが,この現象でもストレスファイバー形成が伴っており,しかも接着斑形成およびRho/ROCK活性化も関わることが明らかになっている103).HUVECでは6種類のLPA受容体の中でLPA6 mRNAの発現が最も高い10, 104).RenらはLPA6に対するsiRNAをHPAECに導入して,LPAによる単層HPAECシートの透過性亢進がおもにLPA6を介して起こることを示した78).Yukiuraらも同様の結果をHUVECで報告しているが,併せてG13–RhoA–Rockシグナル経路によるストレスファイバー形成が透過性亢進に関わることも示している104).細胞外のLPAを分解するエクト酵素である脂質リン酸ホスファターゼ3(LPP3)は細胞膜上に発現しており,細胞周囲のLPA濃度を低く維持するのに寄与している.彼らはさらに,HUVECにおいてLPP3は細胞間接着部位の細胞膜上にもっぱら発現し,非接着部位の細胞膜上にはほとんど発現がないことを見いだした104).この発現パターンと一致して,LPA6を介したLPA刺激が形成するストレスファイバーは,他の細胞と接していない部分の膜直下に局在していた.血管のECはほとんどが他のECと相互に接着していると考えられることから,ECは血中のLPAによりバリア機能が損なわれることをLPP3の発現を通して普段から防いでいるらしい.また,血管が新生する際のECでは,新生血管の先端部に細胞と接しない部位が現れるので,LPAはこの先端部で血管新生に何らかの作用を発揮していることも考えられる.
11)毛髪成長とLPA6
毛髪の成長の場である毛包に対するLPAの効果は,2003年にLPAがホスファチジン酸とともに毛包上皮細胞の増殖を促進することを報告した論文に記載がある105).ただし,LPAの毛包上皮細胞に対する増殖促進効果はホスファチジン酸よりもかなり低い.2006年にKazantsevaらは連鎖解析によって,2-アシルLPAを産生する膜結合型ホスファチジン酸選択的ホスホリパーゼA1の一つであるmPA-PLA1αをコードする遺伝子(LIPH)が常染色体劣性遺伝の乏毛症の原因遺伝子であることを発見した106).その後,二つの独立した研究グループによって,LPA6(論文中ではP2Y5として記載)をコードする遺伝子(P2RY5)のいくつかの変異もまた同様の先天性乏毛症を引き起こすことが報告された9, 29).先天性乏毛症を引き起こす二つの遺伝子の変異は,上記以外にも現在までに多数報告されている107).おそらく報告されてきた変異のほとんどは,遺伝子発現を妨げるかもしくは酵素や受容体としての機能を損なわせる原因になると思われる.mPA-PLA1αおよびLPA6のmRNAはともにヒト毛根の内毛根鞘において発現することから29, 30),mPA-PLA1α由来の2-アシルLPAは,オートクライン/パラクライン様式でLPA6を活性化して毛根を刺激する機序が考えられる.LPA6が1-アシルLPAよりも2-アシルLPAにリガンド選択性を示すこと10)(2-3)項を参照)を考慮すると,この機序は合理的である.
Inoueらは,mPA-PLA1α-KOマウスは明らかな乏毛症の症状は呈さないが,多くの乏毛症患者が合併する縮毛症に似た症状を呈し,波状の奇形毛髪を持っていることを報告した61).マウス毛包における多くのLPA分子種の相対的存在量を調べると,mPA-PLA1α-KOマウスにおいて不飽和脂肪酸を含むLPA種が劇的に減少していた.さらなる解析の結果,減少していたのは1-アシルLPAではなく,主に2-アシルLPA(>90%)であることを確認した.マウス毛包でmPA-PLA1α由来の不飽和2-アシルLPAが最も豊富なLPA種であることは,正常な毛髪成長にmPA-PLA1αおよびLPA6が重要であることを示唆するヒトの研究結果と一致している.彼らはマウス初代培養ケラチノサイト,ヒトHaCaTケラチノサイトおよびHEK293細胞を用いたin vitro実験で,LPA6–G12/13–ROCKシグナル伝達の下流に,毛包発達に重要なTNF-α変換酵素–TGF-α–EGFRの活性化経路が存在することを示した61).in vivoの実験でもmPA-PLA1α-KOマウスは野生型マウスと比較して,毛包におけるTGF-αの量および皮膚におけるEGFRのリン酸化が減少しており,in vitro実験と矛盾のない結果を得ている.
常染色体優性遺伝形式を示す乏毛症が,ケラチン71(K71)をコードする遺伝子の変異によって発症することが報告されている108).このK71変異体にはケラチン中間径フィラメントの形成を阻害する働きがあった.興味深いことにK71タンパク質は,ヒト毛根の内毛根鞘における発現部位がmPA-PLA1αと重複していた.また,mPA-PLA1α-KOマウスの皮膚におけるK71の発現量は,野生型マウスよりも有意に低下していた61).ゆえに,LPA6シグナルはK71の発現調節を介してヒトの正常な毛髪成長に寄与する可能性がある.またK71の発現調節にはEGFRの活性化が介在するかもしれない.