2. 恒常性の破綻によるPAMPsからDAMPsへの変化
a. PRRsによる核酸の認識
植物や動物,ウイルスも含めた核酸に遺伝情報を保存する存在は,自己を定義する形質の複製と保持のため,それぞれ独自のシステムを発展させ,それにより多様な形態の核酸が自然界には存在している.この異なった形態の核酸の存在は自己と非自己を区別するには格好の標的であり,生物はこれらを認識する機構としてPPRsを進化させてきた.2000年の細菌やウイルスに特徴的なCpGモチーフを多く含む非メチル化DNA (CpG-DNA)のTLR9による認識機構の発見を皮切りに4),TLR3やretinoic acid-inducible gene I (RIG-I)やmelanoma differentiation-associated gene 5 (MDA5)による二本鎖RNA(dsRNA)の認識,TLR7によるGU配列を多く含む一本鎖RNAの認識等,宿主にはほとんど存在せず,各種病原体に特徴的な核酸分子に対する特異的な受容体が次々と発見されてきた.さらにはPRRsによるPAMPsの認識について詳細な解析が進むにつれ,PRRsの発現部位が病原体の侵入経路に合わせて適切に制御されていることも明らかとなってきた.例として,TLR3, 7, 9はウイルスや一部の細菌の侵入経路であるエンドソーム内でPAMPsと結合することでMyeloid differentiation primary response 88(Myd88)やTIR-domain-containing adaptor-inducing interferon-β (TRIF)といったアダプタータンパク質を介したシグナルを活性化し,病原体の侵入を迅速に感知していることが明らかとなっている1).加えて,宿主内に存在するさまざまな自己DNAに対しては,細胞質内外に恒常的に存在するDNA分解酵素によりその総量と場所をコントロールすることでPRRsを介した自己核酸の誤認識が起こらないようにしていることも示唆されている5).実際に死細胞から放出される宿主由来のDNAであってもトランスフェクション試薬を用いて細胞内へ導入した場合,強い免疫応答が誘導されるが,DNA切断酵素で処理した場合,その免疫活性化能は失われることがin vitroの実験で証明されている6).これらの研究は自己核酸の影響を最小化することで宿主が病原体由来の“非自己”核酸のPRRsによる認識を最大化していることの傍証として“Strange theory”を支持する一方,細胞死によって生じる自己核酸もPPRsを直接活性化できる証拠として“Danger theory”の一例として多く引用されてきた.
b. アポトーシスの破綻とそれに伴う自己核酸による炎症反応の発見
Kerrらが1972年に核の収縮を伴うヒト組織の細胞死を発見してから約50年,アポトーシス(apoptosis)は最もよく研究された細胞死である.細胞がウイルスや細菌の感染,紫外線による核の損傷等の重度のストレスにさらされることで,システインプロテアーゼである種々のCaspaseが活性化し,細胞内ではエンドヌクレアーゼによる細胞核DNAの断片化,クロマチンや細胞質の凝集が引き起こされ,特徴的な形態的変化を伴ったアポトーシスが誘導される7).当初からKerrが言及したように,このCaspaseによって制御されたアポトーシスは感染細胞の除去や個体の恒常性維持のためにさまざまな臓器で日常的に起こっているにもかかわらず,炎症反応やそれに伴う組織の破壊はほとんど誘導しない.これはアポトーシスを起こした細胞が核内の核酸やミトコンドリアDNA,さらには細胞質内のさまざまなタンパク質を細胞外に放出することなく,速やかにマクロファージによって貪食されるからだと考えられている8, 9).マクロファージに貪食された細胞のタンパク質や核酸はマクロファージのリソソーム内に存在するさまざまな酵素で分解され,新しい細胞の材料となる.特にリソソーム内に存在するDNA分解酵素であるDNase-IIは,その欠損マウスモデルにおいてType-I IFNやIL-6の上昇とそれに伴う無菌性の多発性関節炎や貧血が観察されることから,死細胞の貪食とその後のDNAの分解がアポトーシスの非炎症性細胞死という特徴に重要であることが示唆されている10, 11).このモデルマウスをはじめとして,全身性エリテマトーデス様の症状において,血中に多量のアポトーシス細胞やDNAが存在することは以前から知られていたが5, 12),これらの分解されなかった自己DNAがどのように炎症反応を誘導しているのかについては,近年まで正確には理解されていなかった.当初は自己の核酸が抗DNA抗体やhigh mobility group box protein 1(HMGB-1)等の核酸結合性のタンパク質と結合することでマクロファージや樹状細胞へ取り込まれ,リソソーム内のTLR9を活性化しているものと考えられていたが13, 14),同時にTLR9非依存性の経路の存在も示唆されていた15, 16).2009年,細胞内のDNA認識機構として新たにstimulator of interferon gene(STING)を介する経路が同定されると17),続く2013年には細胞内DNAを基質としてcyclic GMP-AMP(cGAMP)を合成する酵素であるcGASがPPRsとして同定された18).cGASはTLR9と違い,CpGモチーフを含まないDNAも認識することが可能であるため,前述したTLR9非依存的な自己核酸の認識と炎症に重要な役割を担っていることが考えられる.実際にSTINGやcGASの欠損マウスでは前述したDNase-II欠損マウスにおける関節炎が消失することから19, 20),自己核酸のPPRsによる認識が自己免疫疾患において重要であるということを実験的に証明した.さらに最新の研究では,酸化ストレスや紫外線による核膜やミトコンドリア膜の消失に伴う細胞質内への核酸の流出や,HMGB1による細胞質内でのDNA構造の変化等もcGASやTLR9による自己核酸認識を強く誘導することが明らかとなってきている21–23).これらアポトーシス研究に端を発する細胞内核酸の分解による恒常性維持についての理解とPRRs欠損マウスによる自然免疫学の発展は,恒常性が破綻した“危険”な状態においては自己核酸もDAMPsとしてPRRsに認識され,病原体感染と同様に炎症応答を誘導することを明らかにした.
3. 非アポトーシス性細胞死によって生じるDAMPs
a. ネクロプトーシス
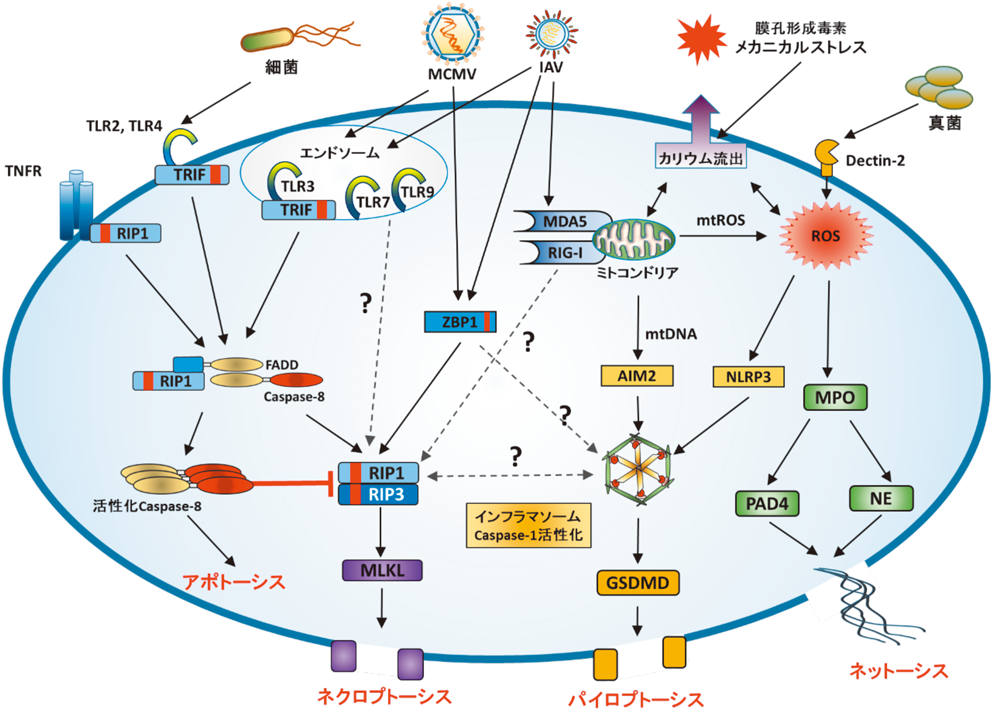
Kerrらによってアポトーシスが報告された翌年,SchweichelとMerkerらは,アポトーシスとは異なった形態学的特徴を示す細胞死が,毒素処理によって誘導されることを報告した.制御されたアポトーシスが細胞膜と核の収縮を特徴とする能動的で“静かな”細胞死であるのに対して,このような細胞膜の破壊によって生じる受動的な細胞死はネクローシス(necrosis)と呼ばれ,それに伴う制御されていない細胞内物質の放出が炎症を誘導するものと長い間考えられてきた.しかし,1988年に腫瘍壊死因子(tumor necrosis factor:TNF)刺激によって,古典的なアポトーシスと同時に核膜の損傷を伴わないネクローシス様細胞死が誘導されることが発見されてから,ネクローシス様細胞死の中にも制御された細胞死が存在するのではないかと考えられてきた.実際にこのTNFによって誘導される膜の溶解を伴う細胞死は,アポトーシスには必須であるCaspaseの抑制下において特に強く誘導されることから24),アポトーシスとは異なったシグナル伝達経路で制御された細胞死であることが明らかであった.2005年にこの制御されたネクローシスの特異的阻害剤としてネクロスタチン-1が発見され25),続いて2008年にはそのターゲット分子としてreceptor-interacting serine/threonine protein kinase 1(RIP1)が同定され26),TNF依存的なネクローシスはキナーゼ依存的な細胞死であることが明らかになった.さらに,続く研究で別のRIPキナーゼファミリー分子としてRIP3が同定され,RIP1とRHIMドメインを介して結合すること27–29),その結合とRIP3のリン酸化がさらに下流のmixed lineage kinase domain-like(MLKL)の活性化と細胞膜の溶解を引き起こすこと等が次々と明らかになり,TNF依存的なネクローシスがアポトーシスとは別のキナーゼの活性化やアダプタータンパク質との複合体の形成を必要とする制御された細胞死であることが証明された30, 31).このような制御された能動的なネクローシス様細胞死はアポトーシスにちなみ,“ネクロプトーシス(necroptosis)”と定義されるようになり,それに伴う細胞内物質の放出も,適切な炎症応答のために厳密に制御されている可能性が示唆された.さらに,TNFによって誘導されるネクロプトーシスとアポトーシスの対比研究は,これら二つの経路がいくつかのタンパク質をアダプタータンパク質として共有していることも明らかにしてきた32).通常,TNFがその受容体であるTNFR1に結合すると,細胞質内でRIP1, TRADD, TRAF2, cIAP, LUBACからなるcomplex Iと呼ばれる複合体を形成する.この複合体はRIP1のユビキチン化を促進し,さらに下流のTAK-1-TAB2-TAB3のシグナルを介して,最終的にNF-κBとMAPKの活性化によるサイトカイン誘導と細胞の生存シグナルを活性化する33, 34).一方,smac模倣薬によるcIAPの阻害やCYLDによるRIP1による脱ユビキチン化によってcomplex Iが不安定な場合,もしくはFasリガンドやTRAILによる刺激によってRIP1, cFLIP, FADD, Caspase-8からなるcomplex IIaを形成した場合には,Caspase-8依存的なアポトーシスが誘導される35).さらにこのcomplex IIaにおいて,cFLIPやCaspase-8が何らかの理由で阻害されている場合,これらによって抑制されていたRIP1, RIP3の活性化に伴うcomplex IIb(またはネクロソーム)と呼ばれる複合体の形成が誘導され,ネクロプトーシスが誘導される36–38).実際にCaspase-8とFADD欠損マウスにおいてはRIP1依存的なネクローシスとそれに伴う炎症や発達不全によって胎生致死の表現型を示すが39, 40),RIP1もしくはRIP3の遺伝子をさらに欠損させることでこれらのマウスの胎生致死の表現型は消失する41, 42).対してRIP1の欠損マウスではRIP3やMLKLの欠損によって回復する全身性の炎症と胎生致死が観察される43–45).さらに発生段階以外でも,RIP3欠損マウスにおいてはTNFによる全身炎症応答が消失し46),薬剤誘導性RIP1欠損マウス47)や腸管や皮膚48, 49)等の組織特異的RIP1欠損マウスにおいてはRIP3依存性ネクロプトーシスの誘導と炎症応答が確認される.これらの遺伝子欠損マウスを用いた研究はRIP1-RIP3を中心としたネクロプトーシスとアポトーシスの相互制御の存在を明らかにし,その破綻がネクロプトーシスによる細胞内物質の放出と炎症反応を誘導しうることを実験的に証明した.
b. 病原体感染によって誘導されるネクロプトーシス
ここまではTNF刺激や遺伝子欠損マウス,Caspase阻害剤によって再現されたネクロプトーシスについて,その細胞死の分子基盤について述べてきた.しかしウイルスや細菌等の病原体の感染によってもアポトーシスと同時にネクローシス様の細胞死が誘導されることは古くから知られている.特にウイルス感染においてはアデノウイルスのE350)やマウスサイトメガロウイルスのvICA等51),さまざまなウイルスタンパク質がCaspase-8を中心としたアポトーシス経路を阻害することが知られており,アポトーシス経路がウイルスの感染成立と宿主防御において中心的な役割を担っていることがわかっていたが,ネクローシス様細胞死についてはウイルス感染がもたらした強いストレスによる受動的な細胞死であるとみなされてきた.しかし前述したように,Caspase-8とRIP1-RIP3の相互的な制御によるネクロプトーシスの存在が明らかになるにつれ,宿主はウイルスの感染によって引き起こされる抗アポトーシス状態を宿主の恒常性を脅かす“危険なもの”としてRIP1-RIP3依存的なネクロプトーシスと細胞内物質の放出,つまりDAMPsによって感知できるように進化してきたものと考えられるようになってきた52).これを裏づけるように,近年では病原体センサーである各種PRRsの刺激がCaspase-8抑制下では強くネクロプトーシスを誘導することが明らかになってきている.例として,ウイルス感染によって生じるdsRNAを認識するTLR3,グラム陰性細菌の細胞壁に存在するリポ多糖(LPS)を認識するTLR4はアダプタータンパク質であるTRIFを介してType-I IFNや各種サイトカイン遺伝子の発現を誘導する分子だと考えられてきたが1),近年の研究によりTRIFに存在するRHIMドメインがpoly(I:C)やLPSの刺激によりRIP1-RIP3との結合を誘導し,Caspase阻害剤の存在下ではネクロプトーシスを強く誘導することが明らかになった53–55).さらに,細胞内のDNAを感知するPRRsと考えられていたZBP1もRHIMドメインを持ち,実際のウイルス感染においてはRIP3を介した細胞死を誘導するセンサー分子として働いていることが最近の研究から明らかとなってきている56–60).興味深いことに,DNAウイルスであるマウスサイトメガロウイルスとRNAウイルスであるインフルエンザウイルスの両方でZBP1依存的な細胞死が確認されているにもかかわらず,同じRNAウイルスである水疱性口内炎ウイルス,さらに紫外線で不活化したウイルスやpoly(I:C),dsDNAではZBP1依存的な細胞死は誘導されない.ZBP1はPKRやADAR1と同様,核酸結合性のあるZαドメインを持ち,その部位が細胞死誘導に重要であることから,ウイルス増殖に伴って生じるdsRNAを感知していると考えられているが,ZBP1とウイルスタンパク質の結合も同様に報告されており,その特異的なリガンドについてはいまだ議論の残る部分である58–60).さらにZBP1やRIP3欠損マウスに対するインフルエンザ感染実験では,用いるインフルエンザ株の違いや,独立したグループの実験でそれぞれ異なる結果が報告されており,その生理学的意義についてもいまだ議論が行われている57, 58, 61, 62).しかし,すでに1型単純ヘルペスウイルスのICP6やマウスサイトメガロウイルスのvIRA等,RHIMドメインを持ち,RIP1-RIP3経路に干渉するウイルスタンパク質も同定されてきていることから,ネクロプトーシスがアポトーシスと同様,宿主防御において重要な役割を担っていることが強く示唆されている63).他のPRRsであるTLR1/2, TLR7, TLR9の刺激によっても非アポトーシス性の細胞死が確認されており55, 64, 65),TRIF-RHIMドメインを介さないネクローシス様細胞死の存在も示唆されている.このようなRHIMドメインを介さないネクロプトーシスに関してはまだ不明な点が多いものの,すでにIFNAR1とIFNGR1によるネクロプトーシスの誘導が報告されていることから66–68),RIG-IやMDA5, STING等の細胞内に存在するその他のPRRsを介したIFNとTNFの分泌も感染細胞やその周囲の細胞をネクロプトーシス感受性にシフトさせている可能性が示唆されている69–71).実際にインフルエンザウイルス感染においてZBP1はType-I IFNによってその遺伝子発現が強く誘導され,それによってネクロプトーシスを亢進させているものと考えられている72, 73).
c. パイロプトーシスとネットーシス
ウイルスや細菌感染によって生じる細胞内dsDNAのabsent in melanoma 2 (AIM2)による認識,細胞外のATPや細胞膜の損傷に伴うカリウム流入や活性酸素種(ROS),それに伴うミトコンドリアの損傷,細菌感染や粒子状物質等のさまざまな刺激によるNACHT, LRR and PYD domains-containing protein 3(NLRP3)の活性化はTLRとは異なった細胞内PRRs経路であり,下流のインフラマソームと呼ばれるタンパク質複合体を介して炎症性Caspaseを活性化する.ヒトではCaspase-1/4/5,マウスにおいてはCaspase-1/11からなるこれら炎症性Caspaseは,LPSやウイルス感染によって誘導された細胞内のpro-IL-1βとpro-IL-18を切断することでこれらのサイトカインの活性化と細胞外への分泌を促し炎症応答を誘導する74).またこの炎症性Caspaseは同時にGSDMDというタンパク質も切断し,切断されたGSDMDが細胞膜の脂質と結合することによって膜の溶解を伴う細胞死も誘導する.これらインフラマソームと炎症性Caspaseの活性化によって誘導される細胞死はパイロプトーシス(pyroptosis)と呼ばれ,ネクロプトーシスと同様,PAMPsの刺激によって誘導される膜の溶解を伴った制御された細胞死である75).この膜の溶解によって,Caspaseによって切断を受けた活性化IL-1βやIL-18のみならず,HMGB1やIL-1α等のさまざまな細胞内物質が放出され,それがDAMPsとなって周辺細胞に働きかけ炎症応答を引き起こす76).
細菌やウイルス感染によって誘導されるその他の細胞死として,核膜の消失と核酸の細胞外への放出を伴うネットーシス(NETosis)についても,ROSによるミエロペルオキシダーゼ(myeloperoxidase:MPO),好中球エラスターゼ(neutrophil elastase:NE)やpeptidyl arginine deiminase, type IV(PAD4)等の細胞内に存在するタンパク質の活性化を必要とする制御された細胞死としてよく研究されている.当初,好中球で報告された細胞外へ放出される核酸はneutrophil extracellular traps(NETs)と呼ばれ,細菌の運動阻害やNETsに付属するカルパインやエラスターセ等のプロテアーゼによる細菌や細菌毒素の無毒化により宿主防御に重要であると考えられていたが,続く研究により放出された核酸を含むこれらの分子が同時にDAMPsとして炎症を誘導していることも明らかになってきた.特にNETsに特徴的なNEやPAD4によって変化したヒストンを含むDNAは細胞内へ取り込まれやすく,TLR9によるサイトカイン分泌を誘導すること,さらにはROSによって酸化されたDNAはDNaseに抵抗性を示し,STING経路をより強く活性化することからもNETosisが核酸DAMPsの放出源としても重要であることが強く示唆されている77).実際のインフルエンザウイルスやセンダイウイルス感染,細菌感染において,DNase処理やNE, PAD4の遺伝子欠損によって炎症症状や組織傷害が低下することからも,NETsによって生じるDAMPsが感染部位の炎症応答の一翼を担っていることが示唆されている.このような病原体感染においてNETsを誘導するPRRsとしては,LPSによるTLR2/4, HIV感染におけるTLR7,細菌の細胞壁由来成分のDectin-2による認識等が報告されているが,それ以外のPRRsの関与や,NETs誘導の詳細な機構についてはまだ不明な点が多い(図1).
PAMPsによって誘導されるネクロプトーシスを含めたこれらの制御された非アポトーシス性細胞死はRIP1/RIP3-MLKL,炎症性Caspase-GSDMD, ROS-MPO/PAD4というようにそれぞれ異なった経路の活性化により膜溶解性の細胞死が引き起こされる.しかし,独立したグループがそれぞれ異なった実験条件で,RIP1-RIP3依存性,非依存性のネットーシス,RIP1やROSによるインフラマソームの活性化,ROSによるRIP1の活性化を伴うネクロプトーシス等を報告しており,これら非アポトーシス性細胞死の正確な分類やその生理学的意味についてはまだ不明な点が多い76, 78, 79).PAMPs以外でもAlumに代表される微小粒子によって誘導される細胞死もDAMPsの放出源として重要であることが知られているが80),特に肺胞マクロファージにおいてRIP3依存的な細胞死がその後の免疫反応に重要であることが我々のグループの研究により明らかとなっており81),臓器や細胞種の違いも細胞死とDAMPsが引き起こす免疫応答にとって重要な要素であると考えられる.さらにはPAMPsが誘導したTNFによるネクロプトーシスの亢進,IFNによるZBP1やNLRP3の発現上昇,膜溶解性によって生じる細胞外HMGB1やIL-1によるネットーシスの誘導82, 83)等も報告されていることから,PAMPsとDAMPsが相互にPRRsシグナルを活性化させることでポジティブフィードバックループを形成し,組織炎症や自己免疫疾患の発症に寄与していることが強く示唆されている(図1,2).
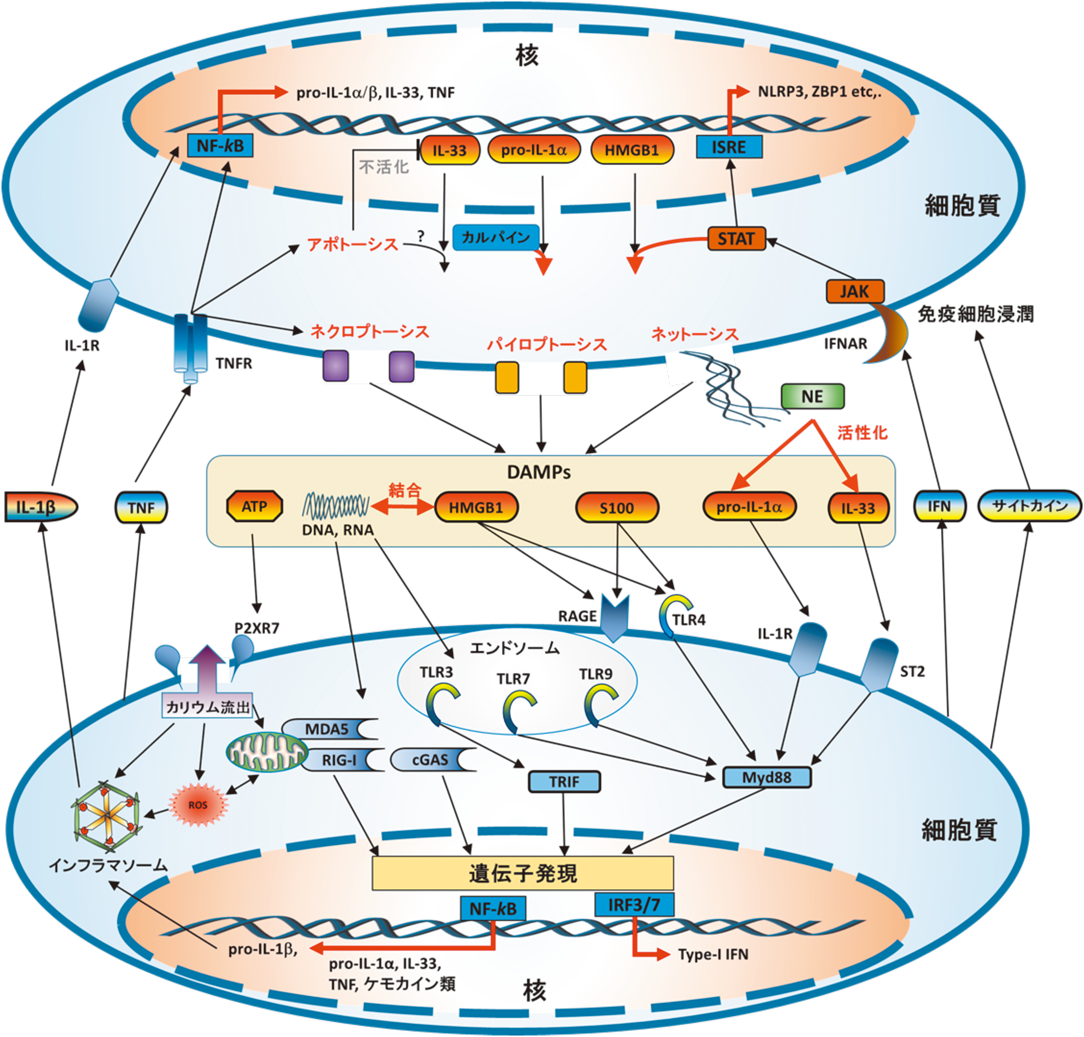
a. 細胞内タンパク質とATP
自己核酸細胞死によって細胞外に放出された核酸がDAMPsとして炎症応答を誘導するのと同様に,いくつかの細胞内タンパク質も細胞外ではDAMPsとして振る舞うことが知られている.その中でもHMGB1はほとんどの細胞で発現しており,その細胞内の機能からDNA結合性も持つため,DAMPsとして特に重要なタンパク質である.HMGB1は核内移行シグナル配列を持ち,核内でDNAシャペロンとしてDNAの安定やさまざまな遺伝子の転写活性を制御している.しかしPAMPsやIFN刺激によるJAK/STAT1経路の活性化はこの核内移行シグナルのアセチル化を誘導し,それによって核内移行シグナルを無効化されたHMGB1は細胞質内にとどまるようになる84).このアセチル化HMGB1を含んだ細胞は,PAMPsやROS,毒素によって強いストレスを受けた場合,パイロプトーシスやネクロプトーシスによる膜溶解によって細胞外に放出される85).細胞外に放出されたHMGB1はRAGEやTLR4と直接結合することにより,NF-κB経路やネットーシスの誘導,DNAとの結合によるTLR9による認識の促進14),CXCL12との結合によるケモカインの安定化86)等を介して,免疫細胞の遊走やサイトカイン分泌,さらなる細胞死を誘導することにより炎症応答を増幅するDAMPsとして振る舞うようになる.事実,敗血症やLPSショックにおける全身性炎症,虚血性再灌流障害,リウマチ等のさまざまなモデルマウスにおいて,HMGB1の中和抗体の投与はその症状を劇的に緩和することから,HMGB1が炎症を増幅するDAMPsとして重要であることが示唆されている87, 88).さらに,細胞内タンパク質であるS100もHMGB1と同様の経路を活性化するDAMPsとして報告されている.現在25種が同定されているS100タンパク質のうち,S100A7, A8, A9とS100A8, A9, A12がそれぞれRAGEとTLR4に特異的に結合するリガンドとしてNF-κBの活性化やサイトカイン分泌,炎症応答やアレルギー疾患への関与が報告されている一方,その他のS100タンパク質がGPCR, EGFRやFGFR1とも結合することも報告されており,これらのDAMPsとしての働きについてはいまだ不明な点が多い89, 90).
タンパク質以外では,細胞内に存在するATPも細胞死によって細胞外に放出され,DAMPsとして振る舞うことが知られている.細胞外に放出されたATPはP2受容体ファミリーの一つであるP2XR7と結合することで細胞膜上のイオンチャンネルが開き,細胞内へのカルシウムの流入とカリウムの流出を引き起こす.この細胞内外のイオンバランスの変化はNLRP3インフラマソームを強く活性化し,IL-1βの切断と活性化を誘導する91).さらに同じP2受容体ファミリーであるP2YR2とATPの結合はアポトーシス細胞の除去や細胞分裂の促進等により障害を受けた組織の治癒を亢進させる一方,好中球や好酸球炎症,IL-33やIL-8による慢性炎症,組織の繊維化,アレルギー疾患の原因となりうることが報告されている92, 93).
b. IL-1ファミリーサイトカイン
通常状態では核内に存在し,細胞外に放出されることでDAMPsとして振る舞うタンパク質としてIL-1αとIL-33がよく研究されている.これらは前述のIL-1β, IL-18とともにIL-1ファミリーサイトカインに属し,免疫細胞の浸潤やサイトカイン増幅による炎症応答の開始と持続に重要なサイトカイン群である.すでにパイロプトーシスの項で述べたように,IL-1βとIL-18はPAMPsによる前駆体の発現誘導と炎症性Caspaseによる切断が必要であるサイトカインであるのに対し,IL-1αは生物活性を持つ前駆体(pro-IL-1α)が細胞内に恒常的に発現しており,細胞死に伴って速やかに放出されるDAMPsとして特に無菌炎症において重要なサイトカインである94).pro-IL-1αにはCaspase-1の切断部位は存在しないものの,膜の溶解を伴う細胞死によって細胞外に放出された後にNK細胞が放出するgranzyme B,マスト細胞のchymase,好中球のNE等,さまざまな細胞由来のタンパク質分解酵素によって切断を受けることでその生物活性がさらに上昇する95).また,定常状態ではpro-IL-1αは核移行シグナルを持ちそのほとんどは核内に存在しているが,細胞質内のカルパインよって切断されることで,核移行配列が切り離され細胞内にとどまるようになる.このカルパインによる細胞内の局在と活性の変化はHMGB1と同様,細胞死に伴うDAMPsとしてのIL-1αの分泌と活性を制御しているものと考えられている.さらに,LPSを代表とするさまざまなPRRsの刺激やTNFにより細胞内のpro-IL-1αの量が増えるのみならず,生物活性を持つ膜結合型IL-1αの細胞表面への発現も著しく上昇することから,無菌性炎症だけではなく病原体感染が誘導する炎症応答においても重要なサイトカインであることが示唆されている96).これらのIL-1αに特徴的な制御機構は,IL-1αがIL-1βと同じ受容体をを介したシグナルを活性化するにも関わらず,生体内では異なった生理活性を示すことをよく説明している97, 98).
IL-33も核移行シグナルとクロマチン結合部位を持つタンパク質で,IL-1αと同様にさまざまな細胞で活性を持った状態で恒常的に発現している.また,分泌シグナルも持たないため,ネクロプトーシスのような細胞死によって受動的に細胞外へと流出すると考えられているが99),ATP刺激によっても細胞外へ分泌されることも報告されているため92),その細胞内局在と放出についてはいまだ不明な点が多い.しかし核移行シグナルを欠損したIL-33の遺伝子導入マウスでは,血中で多くのIL-33が検出され,その受容体であるST2を介した致死性の炎症反応が誘導されることから100),核外に放出されることによって炎症を誘導するDAMPsであると考えられている.IL-33もIL-1αと同様,細胞外へ放出された後,chymaseやNE等,炎症担当細胞が分泌するプロテアーゼで切断されることでその活性が著しく上昇する101).一方,細胞内ではアポトーシスにおいて活性化するCaspase-3, 7によってIL-33の活性部位が切断され,その炎症誘導能は消失する102).IL-33の受容体であるST2の活性化はTH2やT-reg, ILC2細胞の活性化,IL-5, IL-13の分泌誘導等,2型免疫反応を強く誘導することから,特に喘息やアトピー性皮膚炎,乾癬等のアレルギー疾患においてその誘発因子として数多く報告されている103).このようなIL-1αによる好中球の遊走94),ネットーシス由来の各種プロテアーゼによるIL-36を含むIL-1ファミリーサイトカインの切断と活性上昇も報告されており95),この経路においても炎症部位においてポジティブフィードバックループが形成されることを強く示唆している(図2).
引用文献References
1) Takeuchi, O. & Akira, S. (2010) Pattern recognition receptors and inflammation. Cell, 140, 805–820.
2) Matzinger, P. (1994) Tolerance, danger, and the extended family. Annu. Rev. Immunol., 12, 991–1045.
3) Jounai, N., Kobiyama, K., Takeshita, F., & Ishii, K.J. (2012) Recognition of damage-associated molecular patterns related to nucleic acids during inflammation and vaccination. Front. Cell. Infect. Microbiol., 2, 168.
4) Hemmi, H., Takeuchi, O., Kawai, T., Kaisho, T., Sato, S., Sanjo, H., Matsumoto, M., Hoshino, K., Wagner, H., Takeda, K., et al. (2000) A Toll-like receptor recognizes bacterial DNA. Nature, 408, 740–745.
5) Napirei, M., Karsunky, H., Zevnik, B., Stephan, H., Mannherz, H.G., & Möröy, T. (2000) Features of systemic lupus erythematosus in Dnase1-deficient mice. Nat. Genet., 25, 177–181.
6) Ishii, K.J., Suzuki, K., Coban, C., Takeshita, F., Itoh, Y., Matoba, H., Kohn, L.D., & Klinman, D.M. (2001) Genomic DNA released by dying Cells induces the maturation of APCs. J. Immunol., 167, 2602–2607.
7) Elmore, S. (2007) Apoptosis: A review of programmed cell death. Toxicol. Pathol., 35, 495–516.
8) Kurosaka, K., Takahashi, M., Watanabe, N., & Kobayashi, Y. (2003) Silent cleanup of very early apoptotic cells by macrophages. J. Immunol., 171, 4672–4679.
9) Green, D.R., Ferguson, T., Zitvogel, L., & Kroemer, G. (2009) Immunogenic and tolerogenic cell death. Nat. Rev. Immunol., 9, 353–363.
10) Yoshida, H., Okabe, Y., Kawane, K., Fukuyama, H., & Nagata, S. (2004) Lethal anemia caused by interferon-β produced in mouse embryos carrying undigested DNA. Nat. Immunol., 6, 49–56.
11) Kawane, K., Ohtani, M., Miwa, K., Kizawa, T., Kanbara, Y., Yoshioka, Y., Yoshikawa, H., & Nagata, S. (2006) Chronic polyarthritis caused by mammalian DNA that escapes from degradation in macrophages. Nature, 443, 998–1002.
12) Williams, R.C. Jr., Malone, C.C., Meyers, C., Decker, P., & Muller, S. (2001) Detection of nucleosome particles in serum and plasma from patients with systemic lupus erythematosus using monoclonal antibody 4H7. J. Rheumatol., 28, 81–94.
13) Means, T.K., Latz, E., Hayashi, F., Murali, M.R., Golenbock, D.T., & Luster, A.D. (2005) Human lupus autoantibody–DNA complexes activate DCs through cooperation of CD32 and TLR9. J. Clin. Invest., 115, 407–417.
14) Tian, J., Avalos, A.M., Mao, S.Y., Chen, B., Senthil, K., Wu, H., Parroche, P., Drabic, S., Golenbock, D., Sirois, C., et al. (2007) Toll-like receptor 9-dependent activation by DNA-containing immune complexes is mediated by HMGB1 and RAGE. Nat. Immunol., 8, 487–496.
15) Boulé, M.W., Broughton, C., Mackay, F., Akira, S., Marshak-Rothstein, A., & Rifkin, I.R. (2004) Toll-like receptor 9-dependent and -independent dendritic cell activation by chromatin–immunoglobulin G complexes. J. Exp. Med., 199, 1631–1640.
16) Ishii, K.J. & Akira, S. (2006) Innate immune recognition of, and regulation by, DNA. Trends Immunol., 27, 525–532.
17) Ishikawa, H., Ma, Z., & Barber, G.N. (2009) STING regulates intracellular DNA-mediated, type I interferon-dependent innate immunity. Nature, 461, 788–792.
18) Sun, L., Wu, J., Du, F., Chen, X., & Chen, Z.J. (2013) Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the Type-I interferon pathway. Science, 339, 786–791.
19) Ahn, J., Gutman, D., Saijo, S., & Barber, G.N. (2012) STING manifests self DNA-dependent inflammatory disease. Proc. Natl. Acad. Sci. USA, 109, 19386–19391.
20) Gao, D., Li, T., Li, X.D., Chen, X., Li, Q.Z., Wight-Carter, M., & Chen, Z.J. (2015) Activation of cyclic GMP-AMP synthase by self-DNA causes autoimmune diseases. Proc. Natl. Acad. Sci. USA, 112, E5699–E5705.
21) Fang, C., Wei, X., & Wei, Y. (2016) Mitochondrial DNA in the regulation of innate immune responses. Protein Cell, 7, 11–16.
22) Andreeva, L., Hiller, B., Kostrewa, D., Lässig, C., de Oliveira Mann, C.C., Jan Drexler, D., Maiser, A., Gaidt, M., Leonhardt, H., Hornung, V., et al. (2017) cGAS senses long and HMGB/TFAM-bound U-turn DNA by forming protein–DNA ladders. Nature, 549, 394–398.
23) Glück, S., Guey, B., Gulen, M.F., Wolter, K., Kang, T.W., Schmacke, N.A., Bridgeman, A., Rehwinkel, J., Zender, L., & Ablasser, A. (2017) Innate immune sensing of cytosolic chromatin fragments through cGAS promotes senescence. Nat. Cell Biol., 19, 1061–1070.
24) Vercammen, D., Beyaert, R., Denecker, G., Goossens, V., Van Loo, G., Declercq, W., Grooten, J., Fiers, W., & Vandenabeele, P. (1998) Inhibition of caspases increases the sensitivity of L929 cells to necrosis mediated by tumor necrosis factor. J. Exp. Med., 187, 1477–1485.
25) Degterev, A., Huang, Z., Boyce, M., Li, Y., Jagtap, P., Mizushima, N., Cuny, G.D., Mitchison, T.J., Moskowitz, M.A., & Yuan, J. (2005) Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat. Chem. Biol., 1, 112–119.
26) Degterev, A., Hitomi, J., Germscheid, M., Ch’en, I.L., Korkina, O., Teng, X., Abbott, D., Cuny, G.D., Yuan, C., Wagner, G., et al. (2008) Identification of RIP1 kinase as a specific cellular target of necrostatins. Nat. Chem. Biol., 4, 313–321.
27) He, S., Wang, L., Miao, L., Wang, T., Du, F., Zhao, L., & Wang, X. (2009) Receptor interacting protein kinase-3 determines cellular necrotic response to TNF-α. Cell, 137, 1100–1111.
28) Zhang, D.-W., Shao, J., Lin, J., Zhang, N., Lu, B.J., Lin, S.C., Dong, M.Q., & Han, J. (2009) RIP3, an energy metabolism regulator that switches TNF-induced cell death from apoptosis to necrosis. Science, 325, 332–336.
29) Vandenabeele, P., Declercq, W., Van Herreweghe, F., & Vanden Berghe, T. (2010) The role of the kinases RIP1 and RIP3 in TNF-induced necrosis. Sci. Signal., 3, re4.
30) Sun, L., Wang, H., Wang, Z., He, S., Chen, S., Liao, D., Wang, L., Yan, J., Liu, W., Lei, X., et al. (2012) Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of RIP3 kinase. Cell, 148, 213–227.
31) Zhao, J., Jitkaew, S., Cai, Z., Choksi, S., Li, Q., Luo, J., & Liu, Z.G. (2012) Mixed lineage kinase domain-like is a key receptor interacting protein 3 downstream component of TNF-induced necrosis. Proc. Natl. Acad. Sci. USA, 109, 5322–5327.
32) Vandenabeele, P., Galluzzi, L., Vanden Berghe, T., & Kroemer, G. (2010) Molecular mechanisms of necroptosis: An ordered cellular explosion. Nat. Rev. Mol. Cell Biol., 11, 700–714.
33) Silke, J. & Brink, R. (2009) Regulation of TNFRSF and innate immune signalling complexes by TRAFs and cIAPs. Cell Death Differ., 17, 35–45.
34) O’Donnell, M.A., Hase, H., Legarda, D., & Ting, A.T. (2012) NEMO inhibits programmed necrosis in an NFκB-independent manner by restraining RIP1. PLoS ONE, 7, e41238.
35) Tenev, T., Bianchi, K., Darding, M., Broemer, M., Langlais, C., Wallberg, F., Zachariou, A., Lopez, J., MacFarlane, M., Cain, K., et al. (2011) The ripoptosome, a signaling platform that assembles in response to genotoxic stress and loss of IAPs. Mol. Cell, 43, 432–448.
36) O’Donnell, M.A., Perez-Jimenez, E., Oberst, A., Ng, A., Massoumi, R., Xavier, R., Green, D.R., & Ting, A.T. (2011) CASPASE 8 inhibits programmed necrosis by processing CYLD. Nat. Cell Biol., 13, 1437–1442.
37) Lin, Y., Devin, A., Rodriguez, Y., & Liu, Z. (1999) Cleavage of the death domain kinase RIP by Caspase-8 prompts TNF-induced apoptosis. Genes Dev., 13, 2514–2526.
38) Feng, S., Yang, Y., Mei, Y., Ma, L., Zhu, D.E., Hoti, N., Castanares, M., & Wu, M. (2007) Cleavage of RIP3 inactivates its caspase-independent apoptosis pathway by removal of kinase domain. Cell. Signal., 19, 2056–2067.
39) Yeh, W.-C., de la Pompa, J.L., McCurrach, M.E., Shu, H.B., Elia, A.J., Shahinian, A., Ng, M., Wakeham, A., Khoo, W., Mitchell, K., et al. (1998) FADD: Essential for embryo development and signaling from some, but not all, inducers of apoptosis. Science, 279, 1954–1958.
40) Kang, T.-B., Ben-Moshe, T., Varfolomeev, E.E., Pewzner-Jung, Y., Yogev, N., Jurewicz, A., Waisman, A., Brenner, O., Haffner, R., Gustafsson, E., et al. (2004) Caspase-8 serves both apoptotic and nonapoptotic roles. J. Immunol., 173, 2976–2984.
41) Kaiser, W.J., Upton, J.W., Long, A.B., Livingston-Rosanoff, D., Daley-Bauer, L.P., Hakem, R., Caspary, T., & Mocarski, E.S. (2011) RIP3 mediates the embryonic lethality of caspase-8-deficient mice. Nature, 471, 368–372.
42) Zhang, H., Zhou, X., McQuade, T., Li, J., Chan, F.K., & Zhang, J. (2011) Functional complementation between FADD and RIP1 in embryos and lymphocytes. Nature, 471, 373–376.
43) Rickard James, A., O’Donnell, J.A., Evans, J.M., Lalaoui, N., Poh, A.R., Rogers, T., Vince, J.E., Lawlor, K.E., Ninnis, R.L., Anderton, H., et al. (2014) RIPK1 regulates RIPK3-MLKL-driven systemic inflammation and emergency hematopoiesis. Cell, 157, 1175–1188.
44) Dillon Christopher, P., Weinlich, R., Rodriguez, D.A., Cripps, J.G., Quarato, G., Gurung, P., Verbist, K.C., Brewer, T.L., Llambi, F., Gong, Y.N., et al. (2014) RIPK1 blocks early postnatal lethality mediated by caspase-8 and RIPK3. Cell, 157, 1189–1202.
45) Kaiser, W.J., Daley-Bauer, L.P., Thapa, R.J., Mandal, P., Berger, S.B., Huang, C., Sundararajan, A., Guo, H., Roback, L., Speck, S.H., et al. (2014) RIP1 suppresses innate immune necrotic as well as apoptotic cell death during mammalian parturition. Proc. Natl. Acad. Sci. USA, 111, 7753–7758.
46) Duprez, L., Takahashi, N., Van Hauwermeiren, F., Vandendriessche, B., Goossens, V., Vanden Berghe, T., Declercq, W., Libert, C., Cauwels, A., & Vandenabeele, P. (2011) RIP kinase-dependent necrosis drives lethal systemic inflammatory response syndrome. Immunity, 35, 908–918.
47) Roderick, J.E., Hermance, N., Zelic, M., Simmons, M.J., Polykratis, A., Pasparakis, M., & Kelliher, M.A. (2014) Hematopoietic RIPK1 deficiency results in bone marrow failure caused by apoptosis and RIPK3-mediated necroptosis. Proc. Natl. Acad. Sci. USA, 111, 14436–14441.
48) Dannappel, M., Vlantis, K., Kumari, S., Polykratis, A., Kim, C., Wachsmuth, L., Eftychi, C., Lin, J., Corona, T., Hermance, N., et al. (2014) RIPK1 maintains epithelial homeostasis by inhibiting apoptosis and necroptosis. Nature, 513, 90–94.
49) Takahashi, N., Vereecke, L., Bertrand, M.J., Duprez, L., Berger, S.B., Divert, T., Gonçalves, A., Sze, M., Gilbert, B., Kourula, S., et al. (2014) RIPK1 ensures intestinal homeostasis by protecting the epithelium against apoptosis. Nature, 513, 95–99.
50) Chen, P., Tian, J., Kovesdi, I., & Bruder, J.T. (1998) Interaction of the adenovirus 14.7-kDa protein with FLICE inhibits fas ligand-induced apoptosis. J. Biol. Chem., 273, 5815–5820.
51) Skaletskaya, A., Bartle, L.M., Chittenden, T., McCormick, A.L., Mocarski, E.S., & Goldmacher, V.S. (2001) A cytomegalovirus-encoded inhibitor of apoptosis that suppresses caspase-8 activation. Proc. Natl. Acad. Sci. USA, 98, 7829–7834.
52) Mocarski, E.S., Upton, J.W., & Kaiser, W.J. (2012) Viral infection and the evolution of caspase 8-regulated apoptotic and necrotic death pathways. Nat. Rev. Immunol., 12, 79–88.
53) He, S., Liang, Y., Shao, F., & Wang, X. (2011) Toll-like receptors activate programmed necrosis in macrophages through a receptor-interacting kinase-3–mediated pathway. Proc. Natl. Acad. Sci. USA, 108, 20054–20059.
54) Seya, T., Shime, H., Takaki, H., Azuma, M., Oshiumi, H., & Matsumoto, M. (2012) TLR3/TICAM-1 signaling in tumor cell RIP3-dependent necroptosis. OncoImmunology, 1, 917–923.
55) Kaiser, W.J., Sridharan, H., Huang, C., Mandal, P., Upton, J.W., Gough, P.J., Sehon, C.A., Marquis, R.W., Bertin, J., & Mocarski, E.S. (2013) Toll-like receptor 3-mediated necrosis via TRIF, RIP3, and MLKL. J. Biol. Chem., 288, 31268–31279.
56) Upton Jason, W., Kaiser William, J., & Mocarski Edward, S. (2012) DAI/ZBP1/DLM-1 complexes with RIP3 to mediate virus-induced programmed necrosis that is targeted by murine cytomegalovirus vIRA. Cell Host Microbe, 11, 290–297.
57) Kuriakose, T., Man, S.M., Subbarao Malireddi, R.K., Karki, R., Kesavardhana, S., Place, D.E., Neale, G., Vogel, P., & Kanneganti, T.D. (2016) ZBP1/DAI is an innate sensor of influenza virus triggering the NLRP3 inflammasome and programmed cell death pathways. Sci. Immunol., 1, aag2045.
58) Thapa, R.J., Ingram, J.P., Ragan, K.B., Nogusa, S., Boyd, D.F., Benitez, A.A., Sridharan, H., Kosoff, R., Shubina, M., Landsteiner, V.J., et al. (2016) DAI senses influenza a virus genomic RNA and activates RIPK3-dependent cell death. Cell Host Microbe, 20, 674–681.
59) Kesavardhana, S., Kuriakose, T., Guy, C.S., Samir, P., Malireddi, R.K.S., Mishra, A., & Kanneganti, T.D. (2017) ZBP1/DAI ubiquitination and sensing of influenza vRNPs activate programmed cell death. J. Exp. Med., 214, 2217–2229.
60) Maelfait, J., Liverpool, L., Bridgeman, A., Ragan, K.B., Upton, J.W., & Rehwinkel, J. (2017) Sensing of viral and endogenous RNA by ZBP1/DAI induces necroptosis. EMBO J., 36, 2529–2543.
61) Nogusa, S., Thapa, R.J., Dillon, C.P., Liedmann, S., Oguin, T.H. 3rd, Ingram, J.P., Rodriguez, D.A., Kosoff, R., Sharma, S., Sturm, O., et al. (2016) RIPK3 activates parallel pathways of MLKL-driven necroptosis and FADD-mediated apoptosis to protect against influenza a virus. Cell Host Microbe, 20, 13–24.
62) Xu, Y.-L., Tang, H.L., Peng, H.R., Zhao, P., Qi, Z.T., & Wang, W. (2017) RIP3 deficiency ameliorates inflammatory response in mice infected with influenza H7N9 virus infection. Oncotarget, 8, 27715–27724.
63) Mocarski, E.S., Guo, H., & Kaiser, W.J. (2015) Necroptosis: The Trojan horse in cell autonomous antiviral host defense. Virology, 479–480, 160–166.
64) Lalanne, A.I., Moraga, I., Hao, Y., Pereira, J.P., Alves, N.L., Huntington, N.D., Freitas, A.A., Cumano, A., & Vieira, P. (2010) CpG inhibits Pro-B cell expansion through a cathepsin B-dependent mechanism. J. Immunol., 184, 5678–5685.
65) Kim, S.J. & Li, J. (2013) Caspase blockade induces RIP3-mediated programmed necrosis in toll-like receptor-activated microglia. Cell Death &Amp Disease, 4, e716.
66) Thapa, R.J., Nogusa, S., Chen, P., Maki, J.L., Lerro, A., Andrake, M., Rall, G.F., Degterev, A., & Balachandran, S. (2013) Interferon-induced RIP1/RIP3-mediated necrosis requires PKR and is licensed by FADD and caspases. Proc. Natl. Acad. Sci. USA, 110, E3109–E3118.
67) Robinson, N., McComb, S., Mulligan, R., Dudani, R., Krishnan, L., & Sad, S. (2012) Type I interferon induces necroptosis in macrophages during infection with Salmonella enterica serovar Typhimurium. Nat. Immunol., 13, 954–962.
68) McComb, S., Cessford, E., Alturki, N.A., Joseph, J., Shutinoski, B., Startek, J.B., Gamero, A.M., Mossman, K.L., & Sad, S. (2014) Type-I interferon signaling through ISGF3 complex is required for sustained Rip3 activation and necroptosis in macrophages. Proc. Natl. Acad. Sci. USA, 111, E3206–E3213.
69) Brault, M., Stetson, D.B., & Oberst, A. (2016) Type I interferon and TNF signaling plays key synergistic role in programmed necrosis. J. Immunol., 196(1 Supplement): 202.217.
70) Schock, S.N., Chandra, N.V., Sun, Y., Irie, T., Kitagawa, Y., Gotoh, B., Coscoy, L., & Winoto, A. (2017) Induction of necroptotic cell death by viral activation of the RIG-I or STING pathway. Cell Death Differ., 24, 615–625.
71) Brault, M., Olsen, T.M., Martinez, J., Stetson, D.B., & Oberst, A. (2018) Intracellular nucleic acid sensing triggers necroptosis through synergistic Type I IFN and TNF signaling. J. Immunol.
72) Kuriakose, T., Man, S.M., Subbarao Malireddi, R.K., Karki, R., Kesavardhana, S., Place, D.E., Neale, G., Vogel, P., & Kanneganti, T.D. (2016) ZBP1/DAI is an innate sensor of influenza virus triggering the NLRP3 inflammasome and programmed cell death pathways. Sci. Immunol., 1, aag2045.
73) Kuriakose, T., Zheng, M., Neale, G., & Kanneganti, T.-D. (2018) IRF1 is a transcriptional regulator of ZBP1 promoting NLRP3 inflammasome activation and cell death during influenza virus infection. J. Immunol., 200, 1489–1495.
74) He, Y., Hara, H., & Núñez, G. (2016) Mechanism and regulation of NLRP3 inflammasome activation. Trends Biochem. Sci., 41, 1012–1021.
75) Shi, J., Zhao, Y., Wang, K., Shi, X., Wang, Y., Huang, H., Zhuang, Y., Cai, T., Wang, F., & Shao, F. (2015) Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature, 526, 660–665.
76) de Vasconcelos, N.M., Van Opdenbosch, N., & Lamkanfi, M. (2016) Inflammasomes as polyvalent cell death platforms. Cell. Mol. Life Sci., 73, 2335–2347.
77) Gehrke, N., Mertens, C., Zillinger, T., Wenzel, J., Bald, T., Zahn, S., Tüting, T., Hartmann, G., & Barchet, W. (2013) Oxidative damage of DNA confers resistance to cytosolic nuclease TREX1 degradation and potentiates STING-dependent immune sensing. Immunity, 39, 482–495.
78) Pasparakis, M. & Vandenabeele, P. (2015) Necroptosis and its role in inflammation. Nature, 517, 311–320.
79) Desai, J., Mulay, S.R., Nakazawa, D., & Anders, H.-J. (2016) Matters of life and death. How neutrophils die or survive along NET release and is “NETosis” = necroptosis? Cell. Mol. Life Sci., 73, 2211–2219.
80) Marichal, T., Ohata, K., Bedoret, D., Mesnil, C., Sabatel, C., Kobiyama, K., Lekeux, P., Coban, C., Akira, S., Ishii, K.J., et al. (2011) DNA released from dying host cells mediates aluminum adjuvant activity. Nat. Med., 17, 996–1002.
81) Kuroda, E., Ozasa, K., Temizoz, B., Ohata, K., Koo, C.X., Kanuma, T., Kusakabe, T., Kobari, S., Horie, M., Morimoto, Y., et al. (2016) Inhaled fine particles induce alveolar macrophage death and interleukin-1α release to promote inducible bronchus-associated lymphoid tissue formation. Immunity, 45, 1299–1310.
82) Meher, A.K., Spinosa, M., Davis, J.P., Pope, N., Laubach, V.E., Su, G., Serbulea, V., Leitinger, N., Ailawadi, G., & Upchurch, G.R. Jr. (2018) Novel role of IL (Interleukin)-1β in neutrophil extracellular trap formation and abdominal aortic aneurysms. Arterioscler. Thromb. Vasc. Biol., 38, 843–853.
83) Papayannopoulos, V. (2017) Neutrophil extracellular traps in immunity and disease. Nat. Rev. Immunol., 18, 134–147.
84) Lu, B., Antoine, D.J., Kwan, K., Lundbäck, P., Wähämaa, H., Schierbeck, H., Robinson, M., Van Zoelen, M.A., Yang, H., Li, J., et al. (2014) JAK/STAT1 signaling promotes HMGB1 hyperacetylation and nuclear translocation. Proc. Natl. Acad. Sci. USA, 111, 3068–3073.
85) Lu, B., Nakamura, T., Inouye, K., Li, J., Tang, Y., Lundbäck, P., Valdes-Ferrer, S.I., Olofsson, P.S., Kalb, T., Roth, J., et al. (2012) Novel role of PKR in inflammasome activation and HMGB1 release. Nature, 488, 670–674.
86) Schiraldi, M., Raucci, A., Muñoz, L.M., Livoti, E., Celona, B., Venereau, E., Apuzzo, T., De Marchis, F., Pedotti, M., Bachi, A., et al. (2012) HMGB1 promotes recruitment of inflammatory cells to damaged tissues by forming a complex with CXCL12 and signaling via CXCR4. J. Exp. Med., 209, 551–563.
87) Ugrinova, I. & Pasheva, E. (2017) Chapter two-HMGB1 protein: A therapeutic target inside and outside the cell. Advances in Protein Chemistry and Structural Biology, ed Donev R (Academic Press), Vol 107, pp 3–76.
88) Lu, B., Wang, C., Wang, M., Li, W., Chen, F., Tracey, K.J., & Wang, H. (2014) Molecular mechanism and therapeutic Modulation of HMGB1 release and action: An updated review. Expert Rev. Clin. Immunol., 10, 713–727.
89) Xia, C., Braunstein, Z., Toomey, A.C., Zhong, J., & Rao, X. (2017) S100 proteins as an important regulator of macrophage inflammation. Front. Immunol., 8, 1908.
90) Donato, R., Cannon, B.R., Sorci, G., Riuzzi, F., Hsu, K., Weber, D.J., & Geczy, C.L. (2013) Functions of S100 proteins. Curr. Mol. Med., 13, 24–57.
91) Couillin, I., Gombault, A., & Baron, L. (2013) ATP release and purinergic signaling in NLRP3 inflammasome activation. Front. Immunol., 3, 414.
92) Kouzaki, H., Iijima, K., Kobayashi, T., O’Grady, S.M., & Kita, H. (2011) The danger signal, extracellular ATP, is a sensor for an airborne allergen and triggers IL-33 release and innate Th2-Type responses. J. Immunol., 186, 4375–4387.
93) Eltzschig, H.K., Sitkovsky, M.V., & Robson, S.C. (2012) Purinergic signaling during inflammation. N. Engl. J. Med., 367, 2322–2333.
94) Chen, C.-J., Kono, H., Golenbock, D., Reed, G., Akira, S., & Rock, K.L. (2007) Identification of a key pathway required for the sterile inflammatory response triggered by dying cells. Nat. Med., 13, 851–856.
95) Clancy, D.M., Sullivan, G.P., Moran, H.B.T., Henry, C.M., Reeves, E.P., McElvaney, N.G., Lavelle, E.C., & Martin, S.J. (2018) Extracellular neutrophil proteases are efficient regulators of IL-1, IL-33, and IL-36 cytokine activity but poor effectors of microbial killing. Cell Reports, 22, 2937–2950.
96) Di Paolo, N.C. & Shayakhmetov, D.M. (2016) Interleukin 1α and the inflammatory process. Nat. Immunol., 17, 906–913.
97) Groß, O., Yazdi, A.S., Thomas, C.J., Masin, M., Heinz, L.X., Guarda, G., Quadroni, M., Drexler, S.K., & Tschopp, J. (2012) Inflammasome activators induce interleukin-1α secretion via distinct pathways with differential requirement for the protease function of caspase-1. Immunity, 36, 388–400.
98) Rider, P., Carmi, Y., Guttman, O., Braiman, A., Cohen, I., Voronov, E., White, M.R., Dinarello, C.A., & Apte, R.N. (2011) IL-1α and IL-1β recruit different myeloid cells and promote different stages of sterile inflammation. J. Immunol., 187, 4835–4843.
99) Rickard, J., O’Donnell, J.A., Evans, J.M., Lalaoui, N., Poh, A.R., Rogers, T., Vince, J.E., Lawlor, K.E., Ninnis, R.L., Anderton, H., et al. (2014) RIPK1 regulates RIPK3-MLKL-driven systemic inflammation and emergency hematopoiesis. Cell, 157, 1175–1188.
100) Bessa, J., Meyer, C.A., de Vera Mudry, M.C., Schlicht, S., Smith, S.H., Iglesias, A., & Cote-Sierra, J. (2014) Altered subcellular localization of IL-33 leads to non-resolving lethal inflammation. J. Autoimmun., 55, 33–41.
101) Lefrançais, E., Roga, S., Gautier, V., Gonzalez-de-Peredo, A., Monsarrat, B., Girard, J.P., & Cayrol, C. (2012) IL-33 is processed into mature bioactive forms by neutrophil elastase and cathepsin G. Proc. Natl. Acad. Sci. USA, 109, 1673–1678.
102) Lüthi, A.U., Cullen, S.P., McNeela, E.A., Duriez, P.J., Afonina, I.S., Sheridan, C., Brumatti, G., Taylor, R.C., Kersse, K., Vandenabeele, P., et al. (2009) Suppression of interleukin-33 bioactivity through proteolysis by apoptotic caspases. Immunity, 31, 84–98.
103) Liew, F.Y., Girard, J.-P., & Turnquist, H.R. (2016) Interleukin-33 in health and disease. Nat. Rev. Immunol., 16, 676–689.