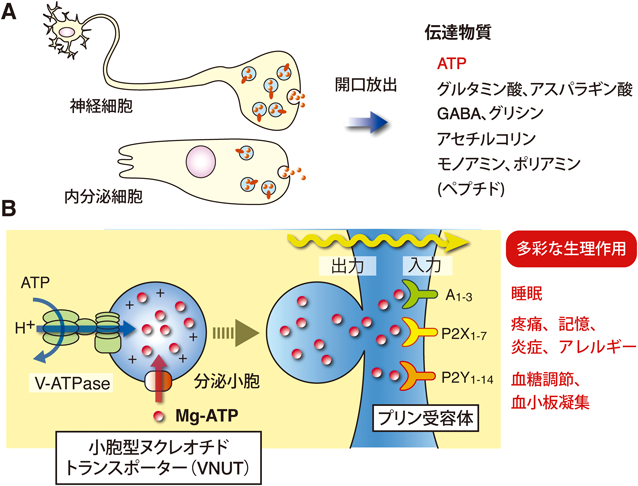
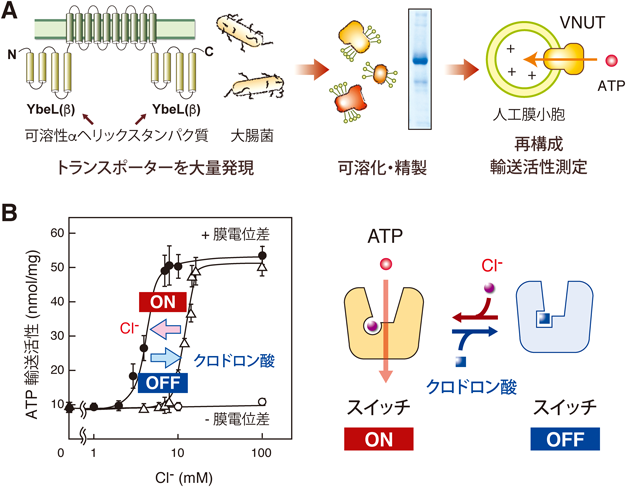
トランスポーターを標的としたプリン作動性化学伝達の特異的遮断薬の同定Identification of a specific inhibitor of purinergic chemical transmission with a focus on transporter
岡山大学自然生命科学研究支援センターAdvanced Science Research Center, Okayama University ◇ 〒700–8530 岡山県岡山市北区津島中1–1–1 ◇ 1–1–1 Tsushima-naka, Kita-ku, Okayama 700–8530
発行日:2018年10月25日Published: October 25, 2018