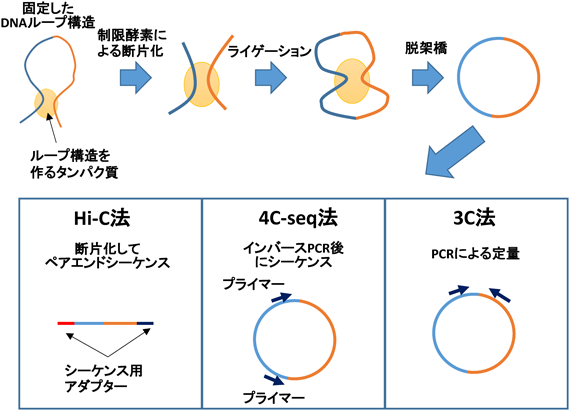
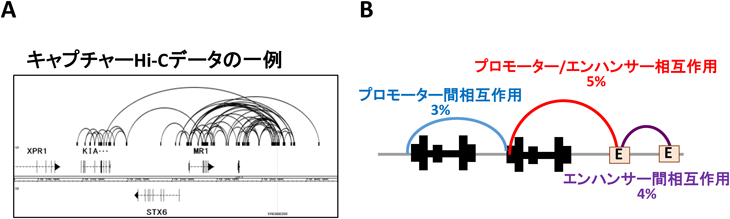
次世代シーケンサーを用いたクロマチン相互作用解析法とその応用Recent development and application of Genome-wide chromatin interaction analysis using NGS
千葉大学大学院医学研究院分子腫瘍学Department of Molecular Oncology, Graduate School of Medicine, Chiba University ◇ 〒260–8670 千葉市中央区亥鼻1–8–1 ◇ 1–8–1 Inohana, Chuo-ku, Chiba 260–8670, Japan
発行日:2018年12月25日Published: December 25, 2018