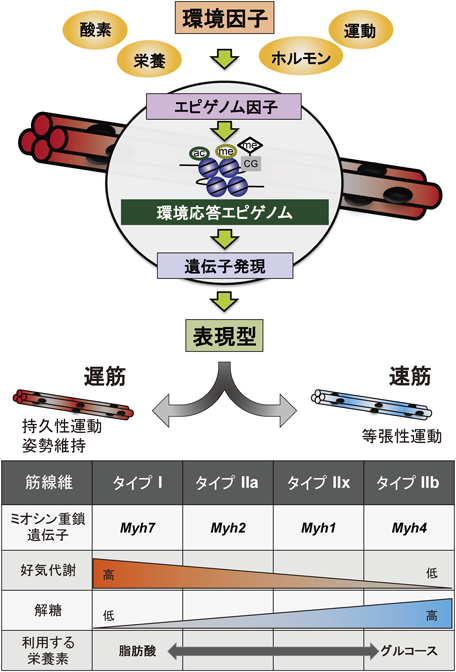
骨格筋は人体重量の40%を占める巨大な臓器であるが,運動器であるとともに,多量のATPを産生・消費する代謝臓器でもある.骨格筋のエネルギー代謝様式は,収縮特性と共役することが知られている.遅筋では,主にミトコンドリア好気代謝によりATPを産生する一方,速筋では,主に解糖系によりATPを産生する.こうした骨格筋の表現型が形成される過程では,エネルギー代謝機能の包括的な転換,すなわち代謝リプログラミングに加え,筋線維遺伝子群の発現を協調させる機構が存在すると考えられるが,その分子機序には不明な点が多い1).
また骨格筋は,栄養環境に応じて,異化と同化のバランスを可塑的に変化させることで,全身の代謝に大きな影響を及ぼす.細胞機能に即した代謝特性を保持しつつ,環境に応じた可塑性を発揮する上で,エピゲノム機構に立脚した代謝制御が存在すると考えられるが,骨格筋の代謝制御におけるエピゲノム機構の役割はほとんど明らかにされていない(図1).
我々は最近の報告で,骨格筋細胞においてヒストン修飾酵素であるリシン特異的脱メチル化酵素-1(lysine-specific demethylase-1:LSD1)が,好気代謝遺伝子群と遅筋線維遺伝子群の発現を協調的に抑制していると同時に,飢餓時に分泌される異化ホルモンであるグルココルチコイドの作用により,LSD1が分解されることを示した2).本稿では,骨格筋の代謝表現型の制御機構について,これまでの知見を概説するとともに,LSD1を中心に,骨格筋の表現型獲得におけるエピゲノム機構の役割について解説する.
本節では,骨格筋の代謝と線維型の制御機構について,これまでに報告された知見を概説する.
1)PGC-1ファミリーによる骨格筋代謝制御
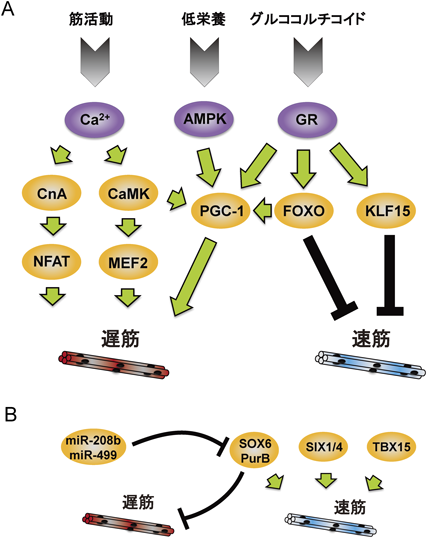
骨格筋における代謝制御において,PGC-1α(peroxisome proliferator-activated receptor gamma, coactivator 1 alpha)が重要な役割を担っており,多くの代謝制御シグナルがこの分子に集約されることが知られている3)(図2A).PGC-1αはもともと褐色脂肪細胞の熱産生を誘導する因子として同定されたが,その後他の代謝臓器でも高発現していることが知られるようになった.PGC-1αを強制発現した骨格筋細胞株では,NRF(nuclear respiratory factor)-1, 2の転写を活性化して,ミトコンドリア関連遺伝子の発現が誘導される4).また,骨格筋のPGC-1αはERRα(estrogen-related receptor alpha)と共役し,血管内皮増殖因子(vascular endothelial growth factor:VEGF)の発現を増加させて血管新生を誘導することが報告されている5).さらに,骨格筋特異的PGC-1αトランスジェニックマウスの骨格筋では,脂肪酸取り込みが増加することが報告されている6).つまり,PGC-1αは骨格筋の好気代謝,血管新生と,代謝基質としての脂肪酸取り込みまでを協調して制御している.
PGC-1ファミリーの筋線維タイプへの関与については,次のような報告がある.まず,骨格筋特異的PGC-1αトランスジェニックマウスの骨格筋では,遅筋線維およびミトコンドリア量が増加しており,持久力が向上していることが報告されている7, 8).一方で,骨格筋特異的PGC-1αノックアウトマウスでは,持久力の著明な低下を来す9)ものの,筋線維タイプは遅筋線維がわずかに減少するのみであった10).また骨格筋特異的PGC-1α/βダブルノックアウトマウスでも,筋持久力の低下はみられるが筋線維タイプには変化がみられなかった11).これらの結果から,PGC-1ファミリーは骨格筋の代謝特性に深く関わるものの,筋線維型の直接的な制御については結論が出ていない.
2)筋活動による筋線維転換
過去の生理学的研究で,遅筋と速筋を支配する運動神経を交換移植すると,遅筋が速筋化し,速筋が遅筋化することが知られている.遅筋と速筋では,支配する神経の電気信号伝達様式が異なっており,遅筋を支配する運動神経は,遅い頻度で長時間持続する電気信号を送るが,速筋では速い頻度で短時間持続する電気信号を送る.また,これらのパターンの電気信号を人工的に運動神経に与えると,遅筋型のパターンでは遅筋型ミオシンの発現を誘導し,速筋型のパターンでは速筋型ミオシンを誘導することが報告されている12).
こうした電気信号パターンの違いは,骨格筋細胞内のカルシウムイオン(Ca2+,以下同)の動態の違いを生む.すなわち,遅筋型の,遅く長時間持続する電気刺激により,Ca2+が低濃度で長時間維持される13).これにより,カルシウム・カルモジュリン依存的セリン−トレオニンホスファターゼであるカルシニューリン(CnA)14, 15)や,カルシウム・カルモジュリン依存的プロテインキナーゼCaMKが活性化すると考えられている16, 17).CnAは転写因子NFAT(nuclear factor of activated T cells)ファミリーを脱リン酸化することにより,これを核に局在化させる18).一方,CaMKは,核内でclass IIヒストン脱アセチル化酵素(HDAC)をリン酸化する.HDACはMEF2(myocyte enhancer factor 2)と核内で複合体を形成してMEF2の転写活性を抑制しており,HDACがリン酸化により核外に排出されることで,MEF2が活性化される16).NFATc1, NFATc3とMEF2は協調して,遅筋関連遺伝子の発現を誘導することが知られている19).また,CaMKは,PGC-1αを活性化し,骨格筋内のミトコンドリア新生や好気代謝を誘導することが知られている20).このように,筋活動と,それによるカルシウムイオン動態の変化は,複数の経路により遅筋線維と好気呼吸を協調して増加させている(図2A).
3)栄養環境による筋線維・代謝制御
摂食や飢餓など,全身の栄養環境の変化に応じて,骨格筋が代謝特性を変化させることはよく知られている.しかし,栄養環境による代謝変化と筋線維を協調させるメカニズムは十分には解明されていない.その中で,飢餓時などに分泌される異化ホルモン,グルココルチコイドが骨格筋に作用すると,グルココルチコイド受容体(GR)を介してPGC-1αや脂肪酸代謝関連遺伝子などを誘導し,好気呼吸を増加させることが知られている21).一方このとき,FOXO(forkhead box-containing protein, O sub-family)1, 3aやKLF15(Krüppel-like factor 15)の誘導を介して速筋選択的に筋萎縮を誘導するため,相対的に遅筋が優位になることが報告されている22, 23)(図2A).
逆に高栄養時と,それを反映したインスリンによる刺激は,骨格筋におけるグルコースの取り込みとグリコーゲンの蓄積を誘導することが知られている24).またインスリンシグナルはFOXO1を不活性化し,下流の好気代謝遺伝子を抑制する.一方でこうしたシグナルが筋線維タイプにどのような影響を及ぼすかについてはほとんど報告がない.
4)速筋型と遅筋型線維の相互排他性
上記のように,遅筋型線維を誘導する環境因子や細胞内シグナルは比較的よく研究されているのに対し,速筋型線維を誘導する環境因子はあまり知られていない.マウスを用いた実験で,SOX625),SIX126, 27),SIX427),TBX1528)などの分子をノックアウトしたマウスの骨格筋で,速筋型線維が減少することが報告されたことから,これらの分子は速筋型を誘導すると考えられている.このうちSOX6は,PurBと協働して遅筋型ミオシン(MYH7, MYH7b)を抑制するが,逆にMYH7, MYH7b遺伝子のイントロン領域にコードされるマイクロRNA, miR-208bとmiR-499が,SOX6とPurBを抑制することが知られている29).つまり遅筋型線維遺伝子の発現が,自身の発現を抑制する転写因子を抑制するという正のフィードバックが形成されており,器官形成における筋線維タイプの相互排他的な選択に大きな影響を与えていると考えられる(図2B).
LSD1は,アミンオキシダーゼファミリーに属する酵素で,タンパク質のメチル化リシン残基を脱メチル化する.主な基質としては,ヒストンH3タンパク質の4番目のリシン残基(H3K4)および9番目のリシン残基(H3K9)の,モノメチル(me1)とジメチル(me2)が知られている30).一般的に,メチル化H3K4は転写活性の高い遺伝子領域に蓄積しており,H3K4me1は活性化エンハンサー,H3K4me2とme3は活性化プロモーターに蓄積している.一方メチル化H3K9は転写が抑制された領域に蓄積している.すなわちLSD1は,ヒストンメチル化の制御を介して遺伝子発現を制御するエピジェネティック因子である.LSD1は,幹細胞の維持や細胞分化,がん化など幅広い生命現象に関与していることが報告されている.我々は先に,LSD1が脂肪細胞やがん細胞においてH3K4の脱メチル化を介して酸化的リン酸化関連遺伝子群を抑制することを示した31, 32).このことからLSD1は細胞のエネルギー代謝と深く関わると考えられるが,骨格筋細胞の代謝におけるLSD1の機能や,栄養環境に応じたLSD1の機能制御については,これまで十分に解明されていなかった.本節では,骨格筋の代謝表現型形成におけるLSD1の役割について,最近我々が報告した内容について解説する.
1)LSD1による遺伝子発現制御
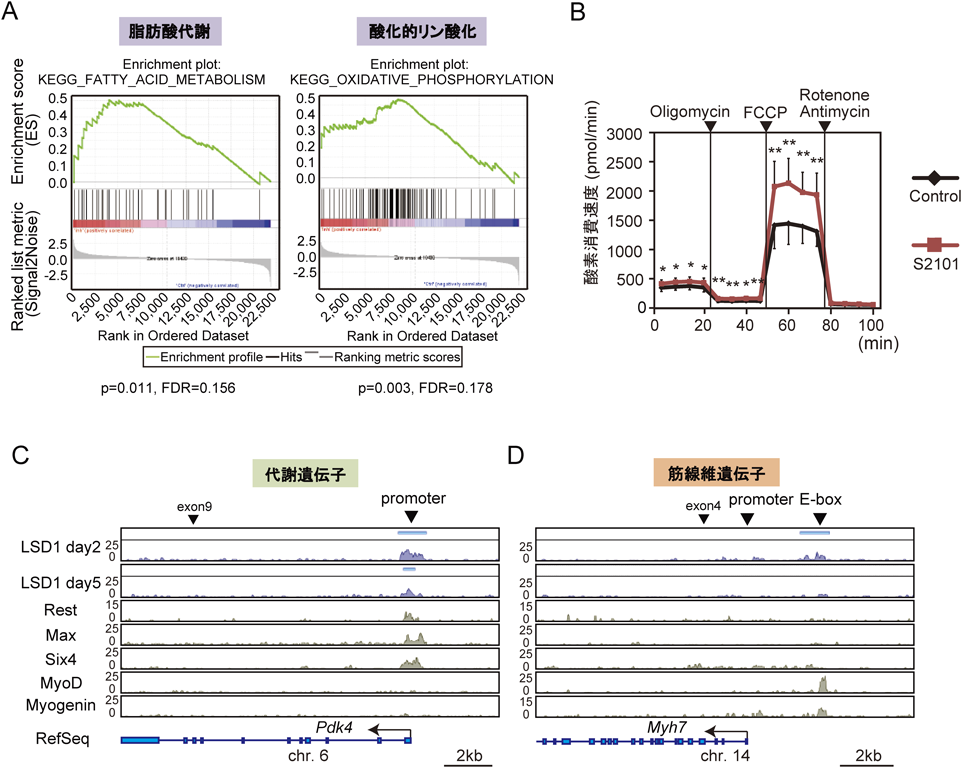
我々は筋分化能を持つマウス骨格筋由来C2C12細胞を用いて,分化過程の遺伝子発現におけるLSD1の役割をマイクロアレイにより解析した.その結果,LSD1阻害下に分化させた細胞では,脂肪酸代謝や酸化的リン酸化に関わる遺伝子群が発現上昇を示した(図3A).またLSD1阻害下に分化させた筋管細胞では,酸素消費量が増加していた(図3B).分化後の細胞で個別の遺伝子発現を解析すると,Pdk4やAtgl, Pgc1αなど好気代謝関連の遺伝子群で発現が亢進していた.一方で,Myh7やMyl2などの遅筋線維遺伝子も,同時に発現上昇を示した.これらの結果から,LSD1は骨格筋の好気代謝と遅筋線維をともに抑制するエピゲノム因子であることが明らかになった.
2)LSD1によるヒストン修飾制御
次に,LSD1が結合して直接遺伝子発現を制御しているゲノム上の領域を同定するために,LSD1抗体を用いたChIP-シークエンス(ChIP-seq)解析を行った.得られたリードデータから,LSD1のピーク検出と遺伝子アノテーションを行い,LSD1集積遺伝子を同定した.先に示した発現マイクロアレイデータと,ChIP-seqとの統合解析を行うと,2種類のLSD1阻害剤でともに1.2倍以上の発現増加がみられる1643遺伝子のうち,70%以上の遺伝子がLSD1集積遺伝子であった.
この結果から,LSD1阻害によって発現が亢進する遺伝子の大部分が,LSD1が発現を制御する直接のターゲットであることが示唆された.実際に好気代謝遺伝子の領域におけるLSD1の分布をChIP-seqデータからみてみると,Pdk4遺伝子では転写開始点(TSS)近傍のプロモーター領域においてLSD1のピークが検出された(図3C).この領域における修飾ヒストンの分布を調べると,H3K4me2とH3K4me3の蓄積がみられた.LSD1阻害によるH3K4メチル化の蓄積の変化を調べたところ,H3K4me2で有意な蓄積の増加があり,H3K4me3でも増加傾向がみられた.AtglやPgc1αのプロモーター領域でも,同様の傾向がみられた.
一方,遅筋線維Myh7遺伝子領域では,TSS近傍ではなく,その約3 kb上流にLSD1の蓄積が検出された.ここには,MyoD1やMyogeninなど筋原性転写因子の結合配列であるE-box配列が存在し,エンハンサー領域であると考えられた(図3D).この領域における修飾ヒストンの分布を調べると,H3K4me1がなだらかに蓄積していた.この領域では,LSD1阻害による有意なH3K4me1の増加はみられなかったが,わずかに増加する傾向がみられた.
以上の結果から,骨格筋細胞の分化過程においてLSD1は,好気代謝関連遺伝子群と,それに共役する遅筋線維遺伝子の領域で,ヒストンH3K4の脱メチル化を介して遺伝子発現を抑制する結果,速筋型の表現型を誘導していると考えられた.
3)グルココルチコイドのLSD1を介した骨格筋表現型制御
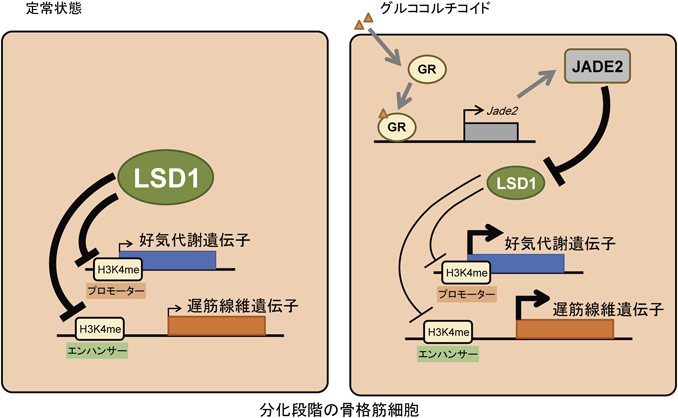
先述のように,骨格筋細胞において,好気代謝と遅筋線維はLSD1の標的遺伝子である.これらの遺伝子群は,飢餓時などに分泌される異化ホルモン,グルココルチコイドの刺激によって,GRを介して誘導されることが知られている.そこで我々は,グルココルチコイドがLSD1の機能に影響を及ぼす可能性を考えた.
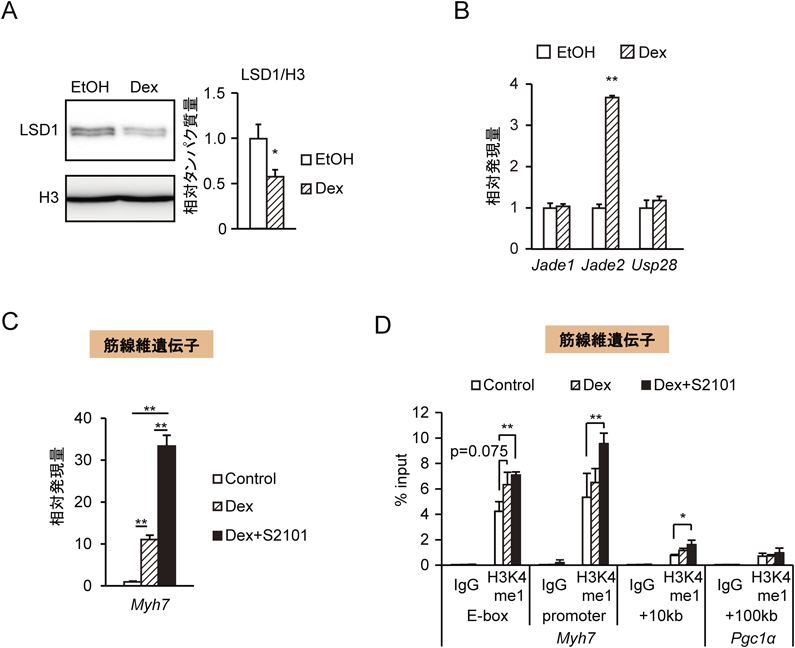
分化後のC2C12細胞を,合成グルココルチコイドであるデキサメサゾン(dexamethasone:Dex)で刺激したところ,興味深いことに,Dex刺激を受けた細胞ではLSD1のタンパク質量が有意に減少していた(図4A).MG132を用いてプロテアソームを阻害するとその減少が緩和されることから,グルココルチコイド刺激がLSD1のプロテアソームでの分解を促進することが明らかになった.そしてC2C12細胞をDexで刺激した際に,E3リガーゼであるJade233)の発現が誘導され(図4B),LSD1の分解に関わることがわかった.
またマウスにDexを腹腔内投与し,腓腹筋を摘出して解析したところ,Jade2の発現が亢進しLSD1のタンパク質量が減少していた.また骨格筋特異的GRノックアウトマウス34)ではこれらの変化がキャンセルされていた.以上の結果から,GRとJade2を介したグルココルチコイドによるLSD1タンパク質の分解は,in vivoの骨格筋でも再現されていた.
先述したように,LSD1は好気代謝と遅筋線維を抑制し,逆にグルココルチコイドはこれらを促進するという,相反する関係にある.そこで次に,C2C12を分化させる際に,DexとLSD1阻害剤を組み合わせて加え,分化後の遺伝子発現を解析した.好気代謝遺伝子のPdk4やPgc1αなどでDexによる発現誘導がみられたが,DexにLSD1阻害を組み合わせると,これらの遺伝子の発現がさらに上昇した.また遅筋線維遺伝子Myh7でも,同様にDexによる誘導と,LSD1阻害を加えたときの増強効果がみられた(図4C).
グルココルチコイドとLSD1阻害剤の組合わせ刺激による,H3K4メチル化の変化をChIP解析で定量した.好気代謝遺伝子のプロモーター領域では,DexとLSD1阻害によりH3K4me2が顕著に増加した.また,遅筋線維遺伝子Myh7のエンハンサー領域とプロモーター領域では,DexとLSD1阻害によりH3K4me1が有意に増加した(図4D).
これらの結果から,グルココルチコイド刺激によってLSD1のタンパク質量が減少し,LSD1の局在部位におけるメチル化H3K4の蓄積を増加させていることが明らかになった(図5).