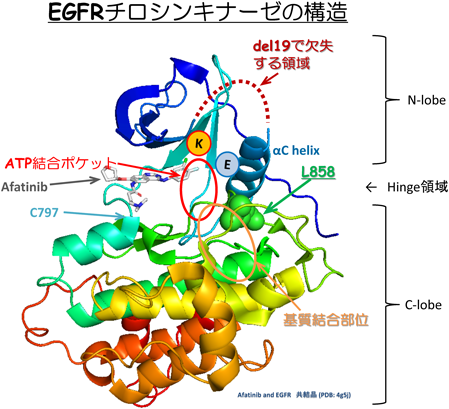
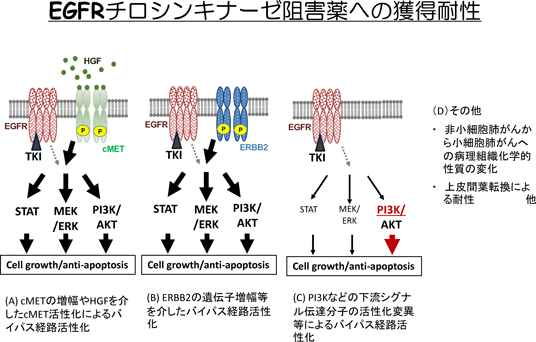
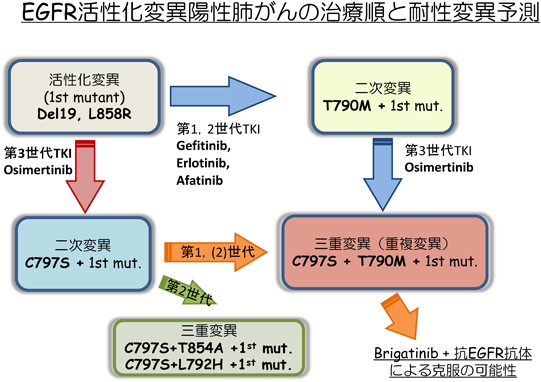
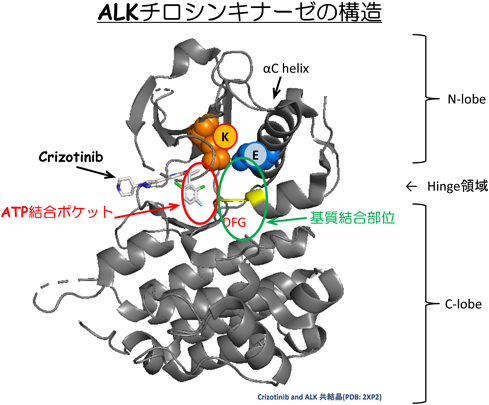
肺がんのがん分子標的薬耐性の基盤Drug resistance mechanisms in non-small cell lung cancer
公益財団法人がん研究会がん化学療法センター基礎研究部Div. of Experimental Chemotherapy, Cancer Chemotherapy Center, Japanese Foundation for Cancer Research ◇ 135–8550 東京都江東区有明3–8–31 ◇ 3–8–31 Ariake, Koto-ku, Tokyo 135–8550, Japan
発行日:2019年2月25日Published: February 25, 2019