多細胞生物の組織内で生じる細胞間の拮抗現象のことを細胞競合(cell competition)と呼ぶ.細胞間の拮抗現象はたとえば細菌類のバクテリオシン,また出芽酵母やゾウリムシのキラー因子などによる異種微生物間の拮抗現象として古くから微生物学においてよく研究されてきた1, 2).その一方で,多細胞生物の組織内における同種細胞間での拮抗作用ということについてはほとんど考えられてこなかった.多細胞生物の体は,通常一つの細胞である卵が細胞分裂によって増殖した多数の細胞から構成されるものであることから,体を構成するすべての細胞は基本的に同じゲノムセットを有する遺伝学的背景が同一の細胞群であると考えられる.しかし実際には,膨大な回数に上る各々の細胞分裂を経る間に生じるDNA複製や染色体分配時のエラー,外界から受けるさまざまなストレス,また細胞呼吸による酸化ストレスのような内在的なストレスも含め,細胞は常にさまざまなエラーや障害を受けつつもそれらを修復しながら生きている.こういったストレスや障害によってDNAがダメージを受け,そのダメージが修復しきれずに変異となって残ってしまった場合,その変異を持つ細胞は遺伝学的背景が周りの正常な細胞とは少しだけ異なってしまうことになる.また,このような実際の遺伝学的変異ではなくても,通常の生理学的条件下において,組織内の同じ起源の細胞が部位特異的もしくは局所的にさまざまな役割を担って各々別の活動を行っているような場合も数多くあり,各々の遺伝子発現や代謝の程度は細胞によって大きく異なっている.つまり,多細胞生物の組織は基本的には同じゲノムセットを有する同一の細胞群から由来するものの,実際にはかなりバリエーションに富んだヘテロな細胞集団からなっている.ではこのヘテロな細胞集団の中で,どのような場合に細胞競合と呼ばれる細胞どうしの拮抗現象が生じるのだろうか.以下に,筆者が実験モデルとしているショウジョウバエでの知見を中心に,細胞競合を通して組織恒常性の維持が行われている一例を紹介したい.
1)Minute変異体
細胞競合という現象は,1975年,当時まだマドリッドのGarcía-Bellido研究室の大学院生だったMorataとRipollによって発見された3).彼らはMinuteと呼ばれるショウジョウバエの変異体を用いて胚発生時の組織の成長と細胞増殖に関する研究を行っていた.このMinuteはリボソームタンパク質をコードする遺伝子群の変異を有しており,古くは1920年代にBridgesとMorganによって,ショウジョウバエの背中の剛毛が野生型に比べて細く短い変異体として,コロンビア大学のいわゆる「モーガンのフライルーム」で発見された4).Minuteのホモ接合変異体(M/M)はリボソームの機能欠損により致死となるが,ヘテロ接合変異体(M/+)は野生型に比べて体表面の剛毛が細く短いことと,胚発生から幼虫を経て成虫になるまでの発生期間が野生型に比べて数日長い(つまり発生速度が少し遅い)という以外には目立った表現型はみられない.MorataとRipollは,このMinute変異体の胚発生期の成長が野生型に比べて遅いのは,体を構成する個々の体細胞の増殖速度が遅いことに起因すると仮説を立て,Minuteヘテロ接合変異体のショウジョウバエの体内に野生型の細胞を作り出して両細胞の増殖速度を比較しようという実験を行っていた.この実験に先立って,彼らの師であったGarcía-Bellidoは,個体の成長速度は細胞の自律的な増殖速度を反映しているものではなく,したがってこれら異なる複数の細胞が同一組織内で異なる速度で増殖するようなことはないだろうと予測した.しかし,実験結果は師の予測が必ずしも正しくないことを示していた.つまり,M/+のショウジョウバエ幼虫の組織に,X線照射による染色体組換えによって野生型の細胞を誘導すると,発生後の成虫では常に野生型細胞の方がM/+変異細胞よりも多く分裂していたことがわかったのだ.一方,ここでさらに興味深いことが判明した.MorataとRipollはさらに,M/+変異細胞の増殖速度が野生型細胞よりも遅いことを証明するために,今度は逆に野生型のショウジョウバエ幼虫の組織にM/+変異細胞を作り出す実験を行った.実験結果はこれまた確かに,M/+変異細胞の増殖速度が野生型細胞よりも遅いことを示していたのだが,彼らはおかしなことに気づいた.この野生型の組織に導入されたM/+変異細胞は,増殖が野生型細胞よりも遅いというだけではなく,成虫になったときには多くのものが消失してしまっていたのだ.そもそもM/+の細胞だけからなるM/+変異体のショウジョウバエは野生型と変わらぬ大きさのハエとして生存可能であることから,M/+変異細胞だけからなるM/+変異個体中ではM/+変異細胞が致死でないことは明らかである.しかし上述の実験から得られた結果は,M/+変異細胞が野生型細胞からなる組織内に存在すると,死んで組織から排除されてしまうことを示していた.このことからMorataとRipollは,増殖速度が異なる2種の細胞が組織内で共存するとそれらの細胞間で競合が起こり,どちらかの細胞が組織内から失われてしまうと結論づけ,この現象を「cell competition」と呼んだ3).ちなみに,上述した彼らの師であったGarcía-Bellidoは自分の仮説が正しくなかったことから,この論文に著者として自分の名前を入れることは辞退し,これが理由でこの歴史的な論文は大学院生2人の名前で出版されることとなったのである5).そしてこの後四半世紀を経た2002年,このMinuteによる細胞競合において正常細胞に隣接するM/+変異細胞が実際にアポトーシスを起こして排除されていることが,当時Morataの学生だったMorenoによって示された6).
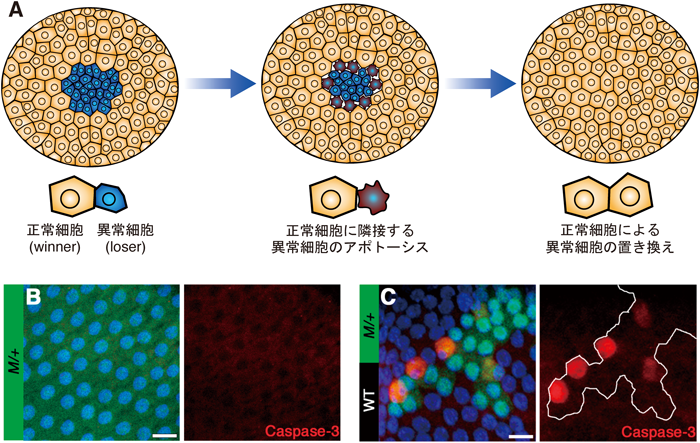
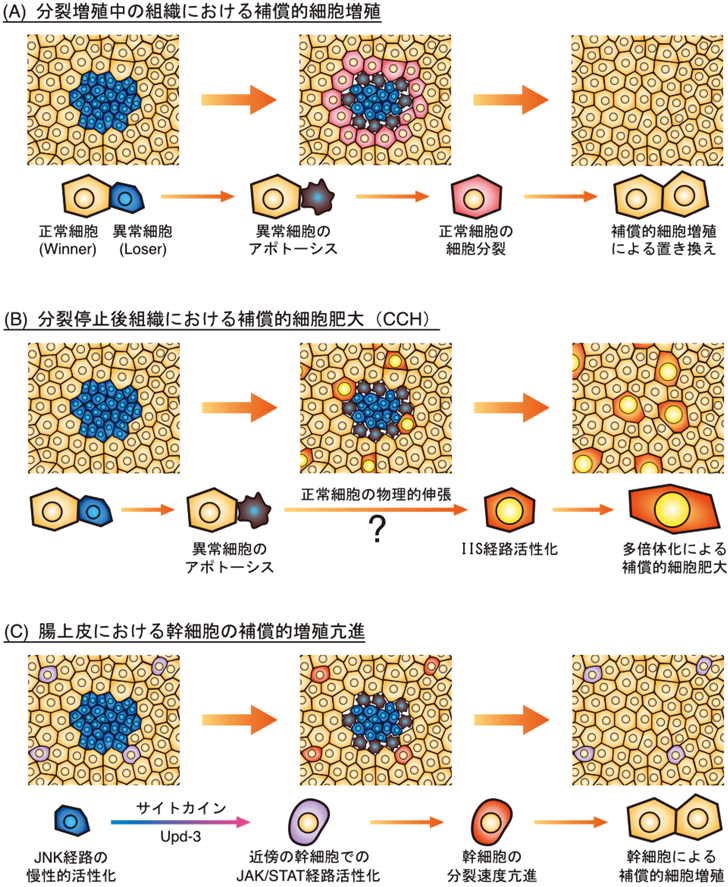
細胞競合という現象の特徴の一つとして,loser cell(敗者細胞)の細胞死があげられる.つまり細胞競合においてloserとなる変異細胞は,基本的にはアポトーシスを起こして組織から除去されていく.細胞競合によって引き起こされるloser cellのアポトーシスは,winner cell(勝者細胞)と隣接しているloser cellにおいて観察される(図1A).これはつまり,細胞競合とは直接的な細胞間相互作用を介して起こっている現象であるということを示している7).このことからloser cellの細胞死は,隣接する正常細胞依存的に起こるものであるということがわかる.これは,たとえばMinute変異細胞(M/+)が組織内で野生型細胞と共存すると細胞死を起こして消えていってしまうのに,Minute変異細胞からなる個体は個体発生が少し遅いだけで,生存も生殖も可能であるということからも明らかである(図1B, C).これらのことから,細胞競合という現象の定義がみえてくる.それはつまり,組織内で異なる2種の細胞が隣接するという状況依存的に“winner”と“loser”が決まり,loserはその組織から排除されるということであり,これは逆にいえば,winnerと隣接していない状況であればloserは生存可能ということになる.ここで「異なる2種の細胞」という書き方をしたが,ではこの細胞競合が起こる原因となる細胞間の違いとは,何の違いだろうか? 細胞たちは互いに何の違いを感じたときに競合し始めるのだろうか?
2)Minuteテクニック
1990年代に分子遺伝学的技術を用いて体細胞分裂時に相同組換えを強制的に誘導する遺伝学的モザイク法が開発されてから8),ショウジョウバエを用いてさまざまな遺伝子に対する変異細胞の表現型が研究されるようになった.それまでは,ホモ接合の個体が胚発生の初期段階で致死になってしまうような変異の表現型を観察することは難しかったが,この遺伝学的モザイク法を用いると,ヘテロ接合個体の正常な組織内にホモ接合の変異細胞を生み出すことが可能になるため,変異体の細胞の表現型を比較的簡単に観察することが可能になった.このようにして正常な組織内にモザイク状に生み出された変異細胞は,多くの場合何らかの表現型,つまり何らかの異常を示す.実際にこの遺伝学的モザイク法を用いて解析されたさまざまな変異細胞の中には,増殖能力が正常細胞に比べて低下していたり,細胞の形態に何かしらの異常が生じていたり,また多くの場合では生存を続けることができず結局は組織からなくなってしまうようなものも多かった7).各々の変異体の報告をみていると,モザイククローンの増殖や生存が低下する原因は,正常細胞との細胞競合による結果ではないかと推察されることが多い.このような中で,正常細胞との細胞競合でloser cellになると報告されている変異細胞の多くに共通する特徴は,増殖速度が正常細胞よりも遅いということである7).一方で,Cyclin D/Cdk4やinsulin/insulin-like growth factorシグナル経路を活性化した変異細胞は,正常細胞に比べて細胞増殖速度が速いにもかかわらず正常細胞との間に細胞競合を起こさない,というような例外もあり9),細胞増殖速度の違いが常に細胞競合の原因となっているわけではないと考えられる.いまだ細胞競合の原因となる変異のタイプがすべて網羅されたわけではないが,リボソームタンパク質に変異を持ったMinute変異細胞のように,代謝や同化といった基本的な細胞の活動状態が正常細胞に比べて低いものがloserになることが多いようである.先述したショウジョウバエの遺伝学的モザイク法を用いて変異細胞の解析を行う研究者にとってはなじみ深い「Minuteテクニック」という実験手法がある10)(図2).たとえば,変異細胞の表現型を解析するにあたって,モザイク法を用いて正常な組織中にホモ接合の変異細胞を作り出した場合,その変異細胞の増殖や生存に対する能力が正常細胞に比べて低いと,変異細胞の細胞数が非常に少なかったり,ある場合はまったくなかったりしてなかなか観察するのが難しいというようなことがしばしば起こる.このようなとき,野生型のバックグラウンドではなく,Minuteヘテロ接合変異体の中に目的のホモ接合変異細胞を作り出す,つまり正常細胞の代わりにMinuteヘテロ接合変異細胞で目的の変異細胞を囲むような状況にすると,比較的大きな変異細胞のクローンを得られて観察を容易にすることが多い.この「Minuteテクニック」として知られている遺伝学的モザイク実験の手法は,まさに細胞競合を利用して,変異細胞クローンに対する周囲の正常細胞による競合のプレッシャーを緩和することによって変異細胞の増殖や生存をレスキューするものである.過去の文献を調べていくと,このMinuteテクニックを用いて変異細胞クローンの増殖をレスキューした例はかなりたくさんあり,変異のタイプもさまざまである7).Minute変異細胞は野生型の細胞に比べて細胞分裂の速度が遅いということに注目すると,このMinuteテクニックでモザイククローンのサイズがレスキューされるという現象はただ単に細胞間の細胞分裂速度の競争のようにもみえるが,分裂速度が遅い変異細胞だけがMinuteテクニックによってレスキューされるわけではないことから,これはただの細胞間の増殖速度の競争ではないということがわかる.
3)がん原遺伝子とスーパーコンペティター
細胞間での発現レベルの差異が細胞競合を引き起こす原因となる代表的な遺伝子としてdMycがある.dMycとは,がん原遺伝子として知られるc-mycのショウジョウバエホモログであり,細胞増殖,細胞周期の制御に重要な役割を持つ転写制御因子である.このdMycは個体の生存に必須な遺伝子であるが,dMycの弱い変異を持つ個体(hypomorphic mutant)は,野生型に比べると成長が不良であるものの生存は可能である.しかし,このhypomorphのdMyc変異細胞をモザイク法によって正常な組織の中に作り出すと,Minuteヘテロ接合変異細胞と同じようにアポトーシスを起こして排除されていく11).同様の細胞競合現象は,がん原遺伝子のRas12, 13),Wingless(Wnt)シグナル経路の構成遺伝子14–16),JAK/STATシグナル経路の構成遺伝子17),そしてHippoシグナル経路の転写共役因子Yki(YAP)18, 19),これらそれぞれの変異細胞と正常細胞との間においても報告されている.ここにあげた細胞競合を引き起こす原因となる変異が関わるシグナル経路は,いずれも細胞増殖の制御に重要な役割を持っており,実際にこれらの変異細胞はすべて,発生中のショウジョウバエ成虫原基上皮組織において増殖速度が正常細胞よりも低いという表現型を示す.また逆に,細胞がこれらのシグナル経路の機能を過剰に活性化させるような変異を持った場合,変異細胞が正常細胞に対してwinnerとなることも発見されており,このような変異細胞をスーパーコンペティターと呼ぶ9, 20).たとえば,dMycを過剰に発現した細胞やYkiが過剰に活性化した細胞を正常細胞からなる成虫原基上皮組織の中に作り出すと,これらの細胞に隣接する周囲の正常細胞が細胞死を起こして組織から排除され,スーパーコンペティターとなった変異細胞は増殖して自らのコピーでどんどん組織を埋めつくしていくことにより,正常細胞にとって代わっていくことになる.同様にして,腸管上皮に生じたAPC(Wntシグナル経路の構成因子)の変異細胞も,周囲の正常細胞にアポトーシスを促しながら増殖してクローン領域を拡大していくことが報告されている16).MycやYkiの活性化,もしくはAPCの変異はいずれもいわゆるがん原性変異であり,これらのように増殖が盛んな変異細胞とはつまりがん細胞になるポテンシャルを持った前がん細胞といえるものである.このような前がん細胞が正常細胞を駆逐しながら組織を占領していくと,さらに二つ目,三つ目のがん原性の変異がヒットする可能性がどんどん高まっていくことになる.これはまさに領域がん化(field cancerization)という概念であり,スーパーコンペティターによる細胞競合はがん初期段階のそれを見事に説明する例としてがん生物学分野から注目を浴びる原因となった21).
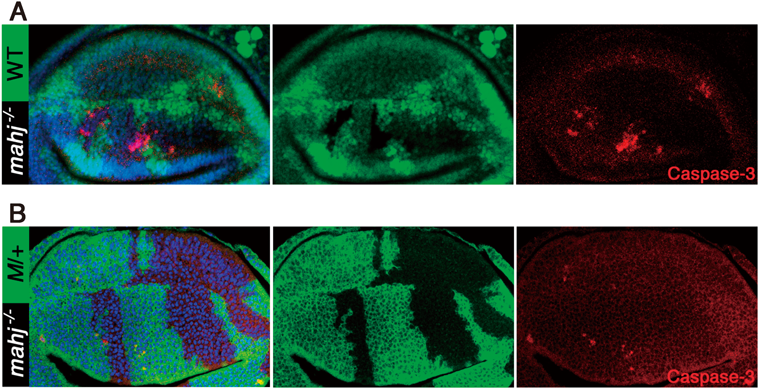
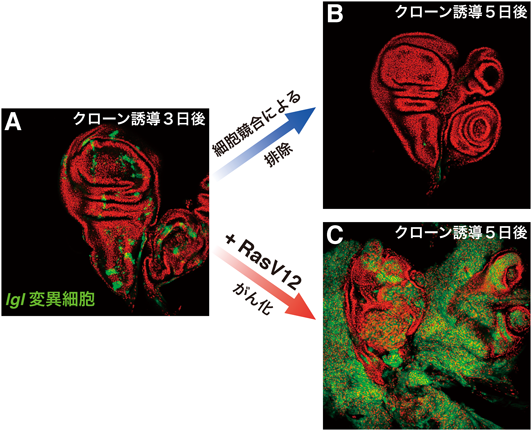
4)細胞極性に関わるがん抑制遺伝子
細胞増殖を制御する遺伝子やシグナル経路以外のもので,もう一つ細胞競合を引き起こす原因となる代表的な変異として,上皮細胞の頂底極性に関わるがん抑制遺伝子(neoplastic tumor suppressor gene:nTSG),lethal giant larvae(lgl),discs large(dlg),scribble(scrib)の変異がある.これら三つの遺伝子の転写産物は,いずれも上皮細胞では側部細胞膜のセプテートジャンクション(哺乳類のタイトジャンクションに相当する)に局在するタンパク質であり,上皮細胞の頂底極性の形成に重要な役割を持っている22).ショウジョウバエにおけるこれらnTSGの変異体はそれぞれ非常によく似た表現型を示す.つまり,胚発生は正常に行われるが,これらの母性タンパク質が枯渇すると幼虫体内の成虫原基はその単層上皮組織の形態を維持できなくなり,分化することができずに過剰増殖を続けて他の成虫原基と融合し,不定形の腫瘍細胞塊になってしまう.この幼虫は蛹化できずに最終的には過剰に成長した幼虫として死に至る.このような上皮組織の崩壊と浸潤性の過増殖といった表現型によって,これら三つの遺伝子は極性形成に関わるがん抑制遺伝子と分類された.しかしながらこれらのホモ接合変異細胞を,遺伝学的モザイク法によって,ヘテロ接合変異体の正常な成虫原基上皮組織内にモザイク状に作り出すと,周りの正常細胞と隣接するものから細胞死を起こして消失していく23–25)(図3A, B).すべての細胞が変異細胞であるホモ接合変異体の上皮組織は腫瘍化して増殖が止まらなくなるのに,正常細胞に囲まれると細胞死を起こすというこの表現型は,まさに細胞競合に典型的なものである.また同様の現象は,哺乳類の細胞を用いた実験においても,Scribをノックダウンした細胞が正常細胞に囲まれると細胞死を起こす現象として確認されている26, 27).正常細胞に囲まれるとloserとなって排除されるこれら変異細胞に,強いがん原性変異として知られる活性化型Ras(RasV12)を発現させると,一転これらは激しい増殖と浸潤を開始し,悪性がんのように振る舞い始める23, 28)(図3C).つまり,nTSGの変異細胞はRasV12を発現することにより,正常細胞との競合を克服したと考えられる.nTSGの変異を持った前がん細胞が,がん原性遺伝子の活性化というさらなる変異を得ることにより悪性化するというこの流れは,多段階発がんという考え方を非常にシンプルな形で表している.一方で,新生がん抑制遺伝子の変異を持たないRasV12だけを発現した細胞は,正常上皮層から押し出されて排除されることがショウジョウバエでも哺乳類の細胞でも観察される29).このRasV12を発現した前がん細胞が上皮層から物理的に押し出されて排除される現象は“extrusion”と呼ばれ,すべての細胞がRasV12を発現した状況であればこのextrusionが起こらないことから,これも細胞競合の一種であると理解されている.さらに最近,このRasV12発現細胞が,マウスの腸管上皮においてもextrusionによって排除されることが筆者らの研究グループによって示されている30).また,Morataの研究グループは,RasV12をlgl変異細胞に強制発現させた場合,lgl変異細胞は過増殖を起こしてがん化するのだが,その場合でも過増殖を起こしているlgl変異細胞クローンが野生型細胞と接している境界ではlgl変異細胞が細胞死を起こしていることを発見した31).またこれらの変異細胞クローンどうしは融合して大きなクローンになっていくことが観察された.つまりこれらのクローンは集まって大きなクローンの塊になることによって野生型細胞との接触を最小限にして細胞競合を逃れているではないかと推測される.実際,融合できなかった小さなクローンは最終的にすべての細胞が排除されてしまうのである.つまり,RasV12はlgl変異細胞の競合能力自体を上昇させたわけではないが,細胞競合によって細胞が失われていくよりも早い速度で細胞を増殖させることができるために競合を克服したと考えられる.一方で,RasV12のみを発現する変異細胞は正常細胞に対してloserになることを考えると,細胞の競合能とはやはり増殖速度だけではなく,何らかの細胞機能が状況依存的に細胞間の差として測られるものなのではないだろうか.
1)ショウジョウバエの卵巣濾胞上皮
このように細胞競合という概念は,ショウジョウバエの成虫原基上皮組織において発見され,そしてそのメカニズムも主にこの発生中の組織で研究されてきたという経緯がある.成虫原基のような増殖中の組織では,loser cellが細胞死を起こし排除されていくと同時に,その排除されたloser cellが存在した場所は周囲のwinner cellが増殖して埋めていくことによって,loser cellがwinner cellで置き換えられていく.つまり細胞競合では,winner cellによるloser cellの“置き換え”が起こることによりloser cellの排除が行われている.そして,発生中の組織においてこのような細胞の大規模な置き換えというイベントが起こった後でも,発生後にでき上がってくる組織のサイズや形は,細胞競合が起こらなかった正常な組織と変わらないということが示されている9).つまり,この細胞競合のプロセスにおいて,loser cellの細胞死とwinner cellの細胞分裂は,組織全体としてのサイズコントロールシステムの中でそのバランスがしっかりと保たれているのである.失われていく細胞(loser cell)を残った細胞(winner cell)が補うことにより全体のサイズを保つというこの現象は,組織修復にも似た組織恒常性維持システムであると考えることができる32).では,発生中,増殖中の組織ではない,つまり細胞分裂が起こっていない状況ではどうだろうか? 細胞の増殖がもはや起こっていない分裂停止後の組織で細胞競合は起こるのだろうか? もしそのような組織の細胞分裂を起こしていない細胞どうしでも細胞競合が起こるのであれば,細胞増殖の速度の違いが細胞競合を起こす要因ではないということになるだけでなく,どのようにして細胞の置き換えが起こるのかという疑問も出てくる.このような背景から,筆者らは細胞競合現象が分裂停止後の成体組織においても起こるのかに着目した解析を行った.この解析を行う上で,ショウジョウバエ卵巣の濾胞上皮組織を実験のモデルとして使用した(図4).この濾胞上皮細胞では細胞周期が卵形成ステージに依存して厳密に制御されているという実験上の利点がある.具体的には,卵形成ステージ1~6の初期においては,通常の有糸分裂サイクルによる体細胞分裂が進行し細胞は増殖しているが,ステージ7に入ると同時にすべての上皮細胞において有糸分裂サイクルはエンドサイクル(endoreplication cycle:内部複製サイクル)にスイッチされる.エンドサイクルは通常の細胞周期とは異なり,M期を飛ばしてS期とG期のみで繰り返される細胞周期であるため,細胞分裂は起こらずDNAの複製によって核の多倍体化が進む.このエンドサイクルは卵形成ステージ7~9の間に3回回ることが知られており,2nだった核は16nまで成長する.ステージ10に入ると,このエンドサイクルも停止し,この後は卵殻形成に必要な遺伝子領域のみを複製する遺伝子増幅サイクルに落ち着く33).少しややこしいようだが,細胞分裂による増殖という点からみると,ステージ1~6では分裂増殖が起こっており,ステージ7以降は細胞増殖が起こっていない分裂停止後組織であるとみることができる.つまりここでは,このステージ7以降の分裂停止後の細胞で細胞競合が起こるのか否かということをまず問うことになる.
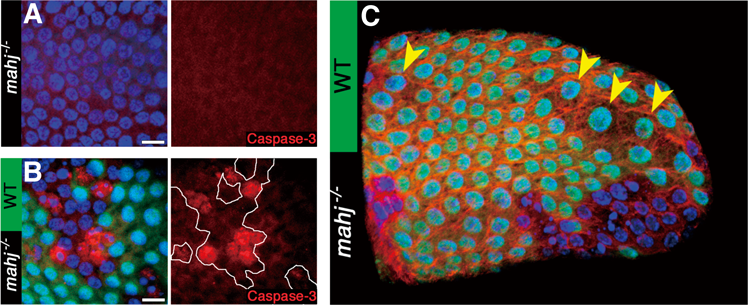
2)分裂停止後組織でのmahjong変異細胞
これを確かめるために,筆者らが細胞競合の制御因子として同定したMahjongの変異細胞によって細胞競合を誘導することを試みた34).Mahjongというのは,筆者らの研究グループが,がん抑制遺伝子nTSGの一つ,lglの変異細胞によって誘導される細胞競合を研究している中で,哺乳類Lglタンパク質(mLgl2)と相互作用を持つ新しいタンパク質として同定したものである25).その後,一連の実験からこのmahjongの変異細胞(mahj)がハエ成虫原基の正常な上皮組織から細胞競合によって排除されること,さらに哺乳類のMDCK培養細胞においてもMahjongをノックダウンした細胞は,正常細胞に囲まれたときにのみ細胞死を起こして単層上皮の頂端側から押し出されて排除されることを発見した25)(図2A).このことはMahjongの機能欠損によって引き起こされる細胞競合がハエから哺乳類まで広く保存された現象であることを示している.他の研究グループからの報告で,MahjongはE3ユビキチンリガーゼ,CRL4DCAF1の基質受容体サブユニットとしての機能を持っており,1000を超えるタンパク質の発現制御に関わっていることが示されている35).つまり,Mahjongが制御している細胞競合能とはかなり多くの種類の遺伝子発現制御による総合的なものであり,競合においてはその相対的な比較がなされているものと考えられる.このように,種を超えて細胞競合を誘導することができるこのmahj変異細胞を,遺伝学的モザイク法を用いて,分裂停止後の野生型卵巣濾胞上皮組織に作り出し,これらmahj変異細胞と野生型細胞間に細胞競合が生じるのかどうかを調べてみたところ,発生中の成虫原基や哺乳類MDCK培養細胞での結果と同様に,細胞競合依存的なmahj変異細胞のアポトーシスが観察された.つまり,この上皮組織のすべての細胞がmahj変異細胞である場合,これらのmahj変異細胞にアポトーシスは観察されなかったが,モザイク状の濾胞上皮で野生型の正常細胞に囲まれたmahj変異細胞はアポトーシスを起こして組織から除去されていった(図5A, B).それまでにショウジョウバエの成虫原基上皮組織で正常細胞との細胞競合を誘導することが報告されていた他のさまざまな変異細胞に関しても調べてみたところ,この分裂停止後の濾胞上皮組織で細胞競合による細胞死が観察されたのは,mahj以外にはMinute変異細胞だけだった.dMycは濾胞上皮細胞ではエンドサイクルの制御に関わっているため,dMyc変異細胞ではエンドサイクルが抑制されて細胞サイズが小さくなり,逆にdMycを強制発現させるとエンドサイクルが亢進されて大きな細胞になる.しかし,どちらの場合も正常細胞との間に細胞競合は観察されなかった34).
3)分裂停止後組織でのnTSG変異細胞
scrib, lgl, dlgといったnTSGの変異細胞についても,卵巣濾胞上皮組織において解析を行ったところ,正常細胞との間にはっきりとした細胞競合を起こすことはなかった34).ステージ6までの分裂期においては,細胞競合によるものと思われる細胞死も少数観察されたが,これらの変異細胞は比較的簡単にこの単層上皮に多層化を誘導して腫瘍を形成した.この卵巣濾胞上皮のような単層の上皮組織は,はっきりとした頂底極性を持った柱状上皮細胞からなっており,この柱状上皮細胞がシート状に並んだ上皮組織としての構造を維持するためには,必ずこのシートの平面に平行な向きに細胞分裂を起こす必要がある36).もし上皮シートの平面に垂直に,つまり上下方向に細胞が分裂してしまうと,シート状の上皮組織の形態が壊れてしまう.ショウジョウバエの上皮組織におけるライブイメージングを用いた詳細な解析から,これらnTSGのタンパク質は,各々がこのような上皮細胞の細胞分裂時に,細胞側面の細胞膜上において分裂装置と相互作用しながら有糸分裂時の紡錘体の向きをシート平面と平行な方向に決定させる役割を果たしていることが明らかにされている37–39).脊椎動物においても,たとえばニワトリの神経上皮組織やヒトのHeLa細胞において,Dlg1が有糸分裂時に上皮細胞の側面に局在することが紡錘体の向きの決定に必要であることが示されている40).また,マウスの乳腺胞において,Scribの欠損が紡錘体の配置異常を引き起こすことにより,管腔内への過形成が生じることも報告されている41).ショウジョウバエの成虫原基でも,scrib, lgl, dlgの変異細胞は,紡錘体の配置が本来の上皮シートに平行な向きから変動してしまうことにより,シート平面と直行する上下方向に分裂することがある.この結果,上皮層内で細胞が上下に積み重なった状態になってしまうが,成虫原基ではこのとき基底膜側に位置する娘細胞が上皮層底部からはがれ落ちてアポトーシスを起こす37).この場合,変異細胞が上皮層底部からはがれ落ちた後にアポトーシスが起こっていることから,積極的にアポトーシスによって排除される細胞競合のプロセスとは異なるものであると考えられるが,どちらにせよこの変異細胞が上皮層から排除されることに変わりはない42).一方で上述したように,ショウジョウバエの卵巣濾胞上皮においては排除されずに腫瘍形成が起こるのはなぜだろうか.これははっきりと実験で証明されたわけではないが,筆者のこれまでの観察から以下のように考えることができる.上で紹介したように,ショウジョウバエの卵巣濾胞上皮細胞の細胞周期は,卵形成ステージに依存して厳密に制御されており,有糸分裂サイクルからエンドサイクルへの移行は,Delta–Notchシグナル経路によって制御されていることがわかっている43, 44).まず,ステージ7の卵の内部にある生殖系列細胞(卵母細胞と哺育細胞)において,リガンドであるDeltaの発現が開始される.濾胞上皮細胞は,頂端側を卵内部へ向けて生殖系列細胞全体を覆っており,生殖系列細胞の細胞膜表面に出てきたDeltaは濾胞上皮細胞の頂端側細胞膜上に存在するNotch受容体と結合する.このDeltaとの結合によって濾胞上皮細胞側のNotchが活性化すると,タンパク質分解酵素によってNotchの細胞質側の細胞内ドメインが切断され,Notch細胞内ドメインは核に移動して一連のNotch応答遺伝子の転写を活性化する.この応答遺伝子の働きによってG2期からM期への遷移を促すCDK(cyclin-dependent kinase)活性の低下,およびG1期からS期への遷移を促すCDK活性の亢進が誘導されることにより,濾胞上皮細胞はエンドサイクルへと移行する.つまり,この有糸分裂サイクルからエンドサイクルへの移行には,卵内部にある生殖系列細胞と外側の濾胞上皮細胞との間での接触依存性シグナル伝達が必要である33).scrib, lgl, dlgの変異細胞が細胞分裂時に紡錘体方向決定の異常により,濾胞上皮のシート平面と直行する上下方向に分裂してしまった場合,基底膜側に位置する娘細胞は生殖系列細胞との接触面を失うこととなり,生殖系列細胞からのリガンド(Delta)を受け取ることができなくなってしまう.この結果,本来ならばステージ7から濾胞上皮細胞内で活性化されるはずのNotchシグナル経路が活性化しないので,この細胞はエンドサイクルへの移行が起こらず,上皮層から逸脱した場所で細胞分裂を継続して行うことになる.scrib, lgl, dlgの変異細胞は上皮頂底極性の形成に不具合が生じているために,上皮層から逸脱した場所で分裂して新たに生まれた変異細胞たちは周りの細胞と組織化された層を形成することができずに腫瘍化するのである.
この分裂停止後の卵巣濾胞上皮組織においてmahj変異細胞が細胞競合によって排除されるという観察結果から,細胞競合は分裂停止後の組織,つまり増殖していない細胞間においても起こることが証明された34).つまり従来考えられてきたように,細胞の増殖速度の違いが細胞競合の原因になる,もしくは増殖速度の速い細胞が遅い細胞を駆逐する,とは必ずしもいえないということが確認された.では細胞たちは何をもって勝ち負けを決めているのか? 上述したようにショウジョウバエではさまざまな異なるタイプの遺伝子に対する変異細胞が正常細胞との間で細胞競合を起こすことが報告されていることから,細胞競合の原因は一つではないと考えられる.なお,分裂停止後の組織における細胞競合現象は筆者らがこの卵巣濾胞上皮で報告した後にも,ショウジョウバエの分裂停止後の網膜組織において余剰の光受容細胞が除去される現象として45),また出生後マウスの心臓において分裂を止めた心筋細胞がMycの発現レベルが低い細胞を除去する現象として46),さらにショウジョウバエの中腸上皮の分化した腸上皮組織においてMinute変異細胞が除去される現象として47),それぞれ報告されている.
1)補償的細胞肥大(CCH)
ところで,細胞分裂が起こっていないステージ7以降の濾胞上皮組織で細胞競合が確認されたことは新たな問いを生み出した.上述したように,細胞競合は組織恒常性維持機構であると考えられ,正常細胞が異常な細胞を排除したのち,補償的な細胞分裂によってその異常細胞の穴埋めをすることも重要なプロセスである.競合という観点から考えると,これは正常細胞(winner)が異常細胞(loser)を自分のコピー(娘細胞)で置き換えていくプロセスであり,これによって組織中の異常細胞はすべて正常細胞に置き換えられるのである.つまりここでの新たな問いとは,細胞分裂を止めた細胞たちが異常細胞を排除したあとにどのようにしてその場を穴埋めするのかということである.当然上皮組織に穴を残しておくことはできないし,実際この分裂停止後の濾胞上皮において細胞競合が起こったあとに穴は観察されない.それでは正常細胞がもう一度有糸分裂サイクルに戻って補償的細胞増殖を起こすのかというと,そうではなかった.この濾胞上皮における分裂停止は,上述したDelta–Notch経路の活性化によって制御される細胞分化を伴っており,ステージ7以降の細胞は細胞分裂を起こせないのである34).ここで筆者らは興味深い現象を発見した.ステージ7以降で細胞競合が起こった場合,いくつかの正常細胞が大きく肥大成長していたのである.しかも,これら肥大成長した細胞は皆,核のサイズが正常のものよりも大きくなっていた(図5C).つまり,これらエンドサイクルステージの正常細胞は,分裂はできないのだがエンドサイクルの速度を速めて多倍体化をさらに促進することによって細胞を肥大成長させていたのである.筆者らは,増殖中の組織でみられる補償的細胞増殖に対して,この現象を補償的細胞肥大(compensatory cellular hypertrophy:CCH)と名づけた34).このCCHに同様な現象は細胞競合の場合だけでなく,ステージ7以降の濾胞上皮に散在的に細胞死を誘導した場合にも確認されたことから,この分裂停止後の上皮における組織修復のシステムであると考えられた.そうはいっても,この濾胞上皮の細胞では内在的にエンドサイクルが進行中であるため,ここでみられた核の多倍体化促進による組織修復システムは,ショウジョウバエの卵巣濾胞上皮に特異的な現象であるとも考えられた.しかしながら非常に興味深いことに,筆者らによるこのCCHの報告の後に,別の研究グループが同じような現象をショウジョウバエ成虫の表皮組織において報告した.成虫の表皮組織は完全に細胞分裂を止めた二倍体(2n)の細胞からなるが,彼らは成虫の腹部表皮を針で刺した傷の治癒過程において,損傷部位周辺の細胞が多倍体化することを発見したのである48).さらに最近,ショウジョウバエの腸管上皮においても,細菌の感染によって上皮がダメージを受けた後の上皮組織修復過程において核の多倍体化が亢進される,つまりCCHによって修復が行われていることが報告された49).またショウジョウバエ以外でも,ヒトを含めた哺乳類や脊椎動物のさまざまな組織において,CCHと同様な現象が相次いで報告されている.たとえば,肝臓は部分切除後に再生することがよく知られているが,この再生過程において多倍体の細胞数が増加することが知られている50).またヒトの角膜内皮においても組織に損傷が生じた場合,もしくは加齢に伴って,肥大した細胞が出現する現象が知られていたが51, 52),マウスの角膜内皮細胞では実際に核の多倍体化が伴っていることが証明された53).ゼブラフィッシュの心臓表面を覆う心外膜組織では,損傷後の再生時に再生が起こっている前縁の細胞でエンドサイクルによる核の多倍体化が起こることにより再生を促していることが発見された54).またつい最近,マウスやヒトの腎臓内の尿細管,そしてマウスの尿路上皮において,損傷後の再生過程で核の多倍体化を伴う細胞肥大が起こっていることが報告されている55, 56).以上のことから,CCHは細胞競合だけでなく組織の損傷修復機構として,分裂停止後の組織において広く保存されているシステムと考えることができる32).
2)補償的細胞成長のメカニズム
ではこのCCHはどのようなメカニズムで起こっているのだろうか.増殖中の組織でみられる補償的細胞増殖については,ショウジョウバエ成虫原基においてそのメカニズムがよく研究されている.成虫原基の上皮組織に散在的なアポトーシスを強制的に誘導すると,その周辺の細胞で細胞分裂が促される.これは,アポトーシスを始めた細胞が周囲の細胞に増殖刺激シグナルを送っていることを示しており,実際アポトーシス経路が活性化した細胞では,Dpp(TGF-β),Wg(Wnt)といった細胞増殖を促進する経路のリガンドが強く発現していること,周辺細胞の補償的細胞増殖がこれらの細胞増殖促進経路の活性化に依存していることが証明されている(図6A)57, 58).しかしながら細胞競合においては,これらの細胞増殖促進経路の関与は確認できないことから,増殖中組織での細胞競合における補償的細胞増殖のメカニズムは,上記のようなアポトーシス誘導性のものとは異なるのではないかと考えられている32).
それでは分裂停止後組織におけるCCHのメカニズムはどうか.まずは増殖中の組織でみられるアポトーシス誘導性の補償的細胞増殖メカニズムと同様のシステムが働いているかどうかを検討した結果,肥大成長している細胞でDpp(TGF-β)やWg(Wnt)といった細胞増殖促進経路の活性化は観察されなかったが,一方でインスリン・インスリン様成長因子シグナル(IIS)経路の関与が確かめられた.また,増殖中組織での補償的細胞増殖とは異なり,CCHはアポトーシスが起こっている細胞の周辺だけでなく,少し離れた場所でも散在的に観察された.さらに,分裂後濾胞上皮にアポトーシスではなくただ単に細胞のサイズが正常のものよりも小さい細胞を作り出した場合にも散在的なCCHが観察された34).以上のことから,分裂後濾胞上皮のCCHではアポトーシスを起こしている細胞が周辺細胞にシグナルを送っているのではなく,数細胞が失われる結果周辺組織に起こる物理的な変化,具体的には細胞にかかる張力の上昇が引き金になっているのではないかと考えている(図6B).この機械的刺激応答によってIISを介した細胞肥大が誘導されるメカニズムに関しては,筆者の研究グループが現在解析中である.
ショウジョウバエの中腸上皮において正常細胞とMinute変異細胞が共存した場合にも,細胞競合によってMinute変異細胞が除去されることが報告されているが47),この腸上皮における例では,分裂増殖している幹細胞だけでなく分化した腸上皮細胞,つまり分裂を停止した細胞間でも細胞競合が起こっている.それではこの腸上皮のように多数の分化した細胞からなる組織中に幹細胞が点在するような組織では,winnerの細胞はどのようにして補償的な組織修復を行うのだろうか.この例においては,CCHのような細胞肥大による組織修復は観察されていないが,その代わりに正常幹細胞の増殖速度が正常時に比べて亢進していることが発見された.組織中のすべての細胞がMinute変異細胞で,競合が起こっていないときでもMinute変異細胞ではストレス応答性のJNK経路が慢性的に活性化していることが以前から知られていたが7),このJNKの慢性的活性化が幹細胞の増殖亢進メカニズムに関与している.つまり,このJNK経路の下流で炎症性サイトカインの一つUpd-3の分泌が促されることにより,このUpd-3を受け取った幹細胞でJAK/STAT経路が活性化することによって分裂速度が高くなるというメカニズムが示されている47)(図6C).
細胞競合とは,組織の中の細胞間で何らかの違いが認識されたときに勝ち負けが決まり,loser cellは組織から排除され,winner cellはloser cellの消失を補うための補償的な行動を起こすことにより置き換えが起こる,というような多段階のプロセスからなっている現象であることを紹介した.細胞が細胞間の違いを認識して勝ち負けを決めるという,非常に重要な第一段階のメカニズムについて考察するために今回,正常細胞との間で細胞競合を起こすことが知られているいくつか異なるタイプの変異細胞について紹介したが,結局細胞間にどのような差異が現れたときに競合が起こるのか,細胞が何を比べることにより勝ち負けが決定するのか,という問いに対して明確な答えを示すことはできていない.しかし実のところ,このようにまだまだわかっていないことが多いというのが細胞競合研究の現状である.一方,紙面の都合上本稿ではほとんどふれなかったが,細胞競合において異常細胞と認識されたloser cellが細胞死を起こすメカニズム,もしくは上皮層から押し出されて排除されるメカニズムに関しては,この10年ほどの間にかなり多くのことがわかってきた印象がある.特に前がん細胞の排除メカニズムに関しては,本誌の2016年第88巻第6号に掲載された井垣の総説に詳しいのでそちらを参照いただきたい.loser cellが組織から除去された後にも,それと同時に一連のプロセスとしてwinner cellによる補償的な細胞成長があり,この段階にも組織修復と器官のサイズ維持という意味で,細胞競合の非常に重要な生理学的意義があることを紹介した.本稿で述べたように,細胞競合は発生中の組織のみならず,分裂停止後の組織を含めた成体のさまざまな組織で起こっていることが示唆されており,この組織修復と器官のサイズ維持に必要な補償的プロセスは,各組織の細胞の性質によってさまざまに柔軟な対応をしているのである.今後,種をまたいでさまざまに性質が異なる組織における細胞競合のメカニズムが解明されることにより,この組織恒常性維持システムの統一的な理解が深まることが期待される.
謝辞Acknowledgments
本稿で紹介した研究は,筆者がFlorida State UniversityのDr. Wu-Min Dengの研究室,および国立遺伝学研究所の鈴木えみ子博士の研究室にて行ったものであり,この場を借りて両研究室の方々に深く感謝する.また,図4の挿絵はDr. Deng研究室のAllison Jevittの厚意により提供いただいた絵を改変したものです.
引用文献References
1) Riley, M.A. & Wertz, J.E. (2002) Bacteriocins:evolution, ecology, and application. Annu. Rev. Microbiol., 56, 117–137.
2) Schmitt, M.J. & Breinig, F. (2006) Yeast viral killer toxins: lethality and self-protection. Nat. Rev. Microbiol., 4, 212–221.
3) Morata, G. & Ripoll, P. (1975) Minutes:mutants of drosophila autonomously affecting cell division rate. Dev. Biol., 42, 211–221.
4) Bridges, C.B., Morgan, T.H., & Washington, C.I.O.(1923) The third-chromosome group of mutant characters of Drosophila melanogaster, Carnegie Institution of Washington. doi:10.5962/bhl.title.24013
5) Díaz, B. & Moreno, E. (2005) The competitive nature of cells. Exp. Cell Res., 306, 317–322.
6) Moreno, E., Basler, K., & Morata, G. (2002) Cells compete for decapentaplegic survival factor to prevent apoptosis in Drosophila wing development. Nature, 416, 755–759.
7) Tamori, Y. & Deng, W.-M. (2011) Cell competition and its implications for development and cancer. J. Genet. Genomics, 38, 483–495.
8) Xu, T. & Rubin, G.M. (1993) Analysis of genetic mosaics in developing and adult Drosophila tissues. Development, 117, 1223–1237.
9) de la Cova, C., Abril, M., Bellosta, P., Gallant, P., & Johnston, L.A. (2004) Drosophila myc regulates organ size by inducing cell competition. Cell, 117, 107–116.
10) Garcia-Bellido, A., Ripoll, P., & Morata, G. (1973) Developmental compartmentalisation of the wing disk of Drosophila. Nat. New Biol., 245, 251–253.
11) Johnston, L.A., Prober, D.A., Edgar, B.A., Eisenman, R.N., & Gallant, P. (1999) Drosophila myc regulates cellular growth during development. Cell, 98, 779–790.
12) Diaz-Benjumea, F.J. & Hafen, E. (1994) The sevenless signalling cassette mediates Drosophila EGF receptor function during epidermal development. Development, 120, 569–578.
13) Prober, D.A. & Edgar, B.A. (2000) Ras1 promotes cellular growth in the Drosophila wing. Cell, 100, 435–446.
14) Giraldez, A.J. & Cohen, S.M. (2003) Wingless and Notch signaling provide cell survival cues and control cell proliferation during wing development. Development, 130, 6533–6543.
15) Vincent, J.-P., Kolahgar, G., Gagliardi, M., & Piddini, E. (2011) Steep differences in wingless signaling trigger Myc-independent competitive cell interactions. Dev. Cell, 21, 366–374.
16) Suijkerbuijk, S.J.E., Kolahgar, G., Kucinski, I., & Piddini, E. (2016) Cell competition drives the growth of intestinal adenomas in Drosophila. Curr. Biol., 26, 1–12.
17) Rodrigues, A.B., Zoranovic, T., Ayala-Camargo, A., Grewal, S., Reyes-Robles, T., Krasny, M., Wu, D.C., Johnston, L.A., & Bach, E.A. (2012) Activated STAT regulates growth and induces competitive interactions independently of Myc, Yorkie, Wingless and ribosome biogenesis. Development, 139, 4051–4061.
18) Neto-Silva, R.M., de Beco, S., & Johnston, L.A. (2010) Evidence for a growth-stabilizing regulatory feedback mechanism between Myc and Yorkie, the Drosophila homolog of Yap. Dev. Cell, 19, 507–520.
19) Ziosi, M., Baena-López, L.A., Grifoni, D., Froldi, F., Pession, A., Garoia, F., Trotta, V., Bellosta, P., Cavicchi, S., & Pession, A. (2010) dMyc functions downstream of Yorkie to promote the supercompetitive behavior of Hippo pathway mutant cells. PLoS Genet., 6, e1001140.
20) Moreno, E. & Basler, K. (2004) dMyc transforms cells into super-competitors. Cell, 117, 117–129.
21) Moreno, E. (2008) Is cell competition relevant to cancer? Nat. Rev. Cancer, 8, 141–147.
22) Bilder, D. (2004) Epithelial polarity and proliferation control: links from the Drosophila neoplastic tumor suppressors. Genes Dev., 18, 1909–1925.
23) Brumby, A.M. & Richardson, H.E. (2003) scribble mutants cooperate with oncogenic Ras or Notch to cause neoplastic overgrowth in Drosophila. EMBO J., 22, 5769–5779.
24) Igaki, T., Pagliarini, R.A., & Xu, T. (2006) Loss of cell polarity drives tumor growth and invasion through JNK Activation in Drosophila. Curr. Biol., 16, 1139–1146.
25) Tamori, Y., Bialucha, C.U., Tian, A.G., Kajita, M., Huang, Y.C., Norman, M., Harrison, N., Poulton, J., Ivanovitch, K., Disch, L., et al. (2010) Involvement of Lgl and Mahjong/VprBP in cell competition. PLoS Biol., 8, e1000422.
26) Norman, M., Wisniewska, K.A., Lawrenson, K., Garcia-Miranda, P., Tada, M., Kajita, M., Mano, H., Ishikawa, S., Ikegawa, M., Shimada, T., et al. (2012) Loss of Scribble causes cell competition in mammalian cells. J. Cell Sci., 125, 59–66.
27) Wagstaff, L., Goschorska, M., Kozyrska, K., Duclos, G., Kucinski, I., Chessel, A., Hampton-O’Neil, L., Bradshaw, C.R., Allen, G.E., Rawlins, E.L., et al. (2016) Mechanical cell competition kills cells via induction of lethal p53 levels. Nat. Commun., 7, 1–14.
28) Pagliarini, R.A. & Xu, T. (2003) A genetic screen in Drosophila for metastatic behavior. Science, 302, 1227–1231.
29) Hogan, C., Dupré-Crochet, S., Norman, M., Kajita, M., Zimmermann, C., Pelling, A.E., Piddini, E., Baena-López, L.A., Vincent, J.P., Itoh, Y., et al. (2009) Characterization of the interface between normal and transformed epithelial cells. Nat. Cell Biol., 11, 460–467.
30) Kon, S., Ishibashi, K., Katoh, H., Kitamoto, S., Shirai, T., Tanaka, S., Kajita, M., Ishikawa, S., Yamauchi, H., Yako, Y., et al. (2017) Cell competition with normal epithelial cells promotes apical extrusion of transformed cells through metabolic changes. Nat. Cell Biol., 125, 1–24.
31) Menéndez, J., Pérez-Garijo, A., Calleja, M., & Morata, G. (2010) A tumor-suppressing mechanism in Drosophila involving cell competition and the Hippo pathway. Proc. Natl. Acad. Sci. USA, 107, 14651–14656.
32) Tamori, Y. & Deng, W.-M. (2014) Compensatory cellular hypertrophy:the other strategy for tissue homeostasis. Trends Cell Biol., 24, 230–237.
33) Klusza, S. & Deng, W.-M. (2011) At the crossroads of differentiation and proliferation: precise control of cell-cycle changes by multiple signaling pathways in Drosophila follicle cells. BioEssays, 33, 124–134.
34) Tamori, Y. & Deng, W.-M. (2013) Tissue repair through cell competition and compensatory cellular hypertrophy in postmitotic epithelia. Dev. Cell, 25, 350–363.
35) Li, W., You, L., Cooper, J., Schiavon, G., Pepe-Caprio, A., Zhou, L., Ishii, R., Giovannini, M., Hanemann, C.O., Long, S.B., et al. (2010) Merlin/NF2 suppresses tumorigenesis by inhibiting the E3 ubiquitin ligase CRL4(DCAF1) in the nucleus. Cell, 140, 477–490.
36) Nakajima, Y.-I. (2018) Mitotic spindle orientation in epithelial homeostasis and plasticity. J. Biochem., 164, 277–284.
37) Nakajima, Y.-I., Meyer, E.J., Kroesen, A., McKinney, S.A., & Gibson, M.C. (2013) Epithelial junctions maintain tissue architecture by directing planar spindle orientation. Nature, 500, 359–362.
38) Bell, G.P., Fletcher, G.C., Brain, R., & Thompson, B.J. (2015) Aurora kinases phosphorylate Lgl to induce mitotic spindle orientation in Drosophila epithelia. Curr. Biol., 25, 61–68.
39) Carvalho, C.A., Moreira, S., Ventura, G., Sunkel, C.E., & Morais-de-Sá, E. (2015) Aurora A triggers Lgl cortical release during symmetric division to control planar spindle orientation. Curr. Biol., 25, 53–60.
40) Saadaoui, M., Machicoane, M., di Pietro, F., Etoc, F., Echard, A., & Morin, X. (2014) Dlg1 controls planar spindle orientation in the neuroepithelium through direct interaction with LGN. J. Cell Biol., 206, 707–717.
41) Godde, N.J., Sheridan, J.M., Smith, L.K., Pearson, H.B., Britt, K.L., Galea, R.C., Yates, L.L., Visvader, J.E., & Humbert, P.O. (2014) Scribble modulates the MAPK/Fra1 pathway to disrupt luminal and ductal integrity and suppress tumour formation in the mammary gland. PLoS Genet., 10, e1004323.
42) Tamori, Y. & Deng, W.-M. (2017) Tissue-intrinsic tumor hotspots: Terroir for tumorigenesis. Trends Cancer, 3, 259–268.
43) Deng, W.M., Althauser, C., & Ruohola-Baker, H. (2001) Notch-Delta signaling induces a transition from mitotic cell cycle to endocycle in Drosophila follicle cells. Development, 128, 4737–4746.
44) López-Schier, H. & St Johnston, D. (2001) Delta signaling from the germ line controls the proliferation and differentiation of the somatic follicle cells during Drosophila oogenesis. Genes Dev., 15, 1393–1405.
45) Merino, M.M., Rhiner, C., Portela, M., & Moreno, E. (2013) ‘Fitness fingerprints’ mediate physiological culling of unwanted neurons in Drosophila. Curr. Biol., 23, 1300–1309.
46) Villa del Campo, C., Clavería, C., Sierra, R., & Torres, M. (2014) Cell competition promotes phenotypically silent cardiomyocyte replacement in the mammalian heart. Cell Rep., 8, 1741–1751.
47) Kolahgar, G., Suijkerbuijk, S.J., Kucinski, I., Poirier, E.Z., Mansour, S., Simons, B.D., & Piddini, E. (2015) Cell competition modifies adult stem cell and tissue population dynamics in a JAK-STAT-dependent manner. Dev. Cell, 34, 297–309.
48) Losick, V.P., Fox, D.T., & Spradling, A.C. (2013) Polyploidization and cell fusion contribute to wound healing in the adult Drosophila epithelium. Curr. Biol., 23, 2224–2232.
49) Xiang, J., Bandura, J., Zhang, P., Jin, Y., Reuter, H., & Edgar, B.A. (2017) EGFR-dependent TOR-independent endocycles support Drosophila gut epithelial regeneration. Nat. Commun., 8, 15125.
50) Miyaoka, Y., Ebato, K., Kato, H., Arakawa, S., Shimizu, S., & Miyajima, A. (2012) Hypertrophy and unconventional cell division of hepatocytes underlie liver regeneration. Curr. Biol., 22, 1166–1175.
51) Ikebe, H., Takamatsu, T., Itoi, M., & Fujita, S. (1986) Age-dependent changes in nuclear DNA content and cell size of presumably normal human corneal endothelium. Exp. Eye Res., 43, 251–258.
52) Ikebe, H., Takamatsu, T., Itoi, M., & Fujita, S. (1988) Changes in nuclear DNA content and cell size of injured human corneal endothelium. Exp. Eye Res., 47, 205–215.
53) Losick, V.P., Jun, A.S., & Spradling, A.C. (2016) Wound-Induced polyploidization: Regulation by Hippo and JNK signaling and conservation in mammals. PLoS One, 11, e0151251.
54) Cao, J., Wang, J., Jackman, C.P., Cox, A.H., Trembley, M.A., Balowski, J.J., Cox, B.D., De Simone, A., Dickson, A.L., Di Talia, S., et al. (2017) Tension creates an endoreplication wavefront that leads regeneration of epicardial tissue. Dev. Cell, 42, 600–615.e4.
55) Lazzeri, E., Angelotti, M.L., Peired, A., Conte, C., Marschner, J.A., Maggi, L., Mazzinghi, B., Lombardi, D., Melica, M.E., Nardi, S., et al. (2018) Endocycle-related tubular cell hypertrophy and progenitor proliferation recover renal function after acute kidney injury. Nat. Commun., 9, 1344.
56) Wang, J., Batourina, E., Schneider, K., Souza, S., Swayne, T., Liu, C., George, C.D., Tate, T., Dan, H., Wiessner, G., et al. (2018) Polyploid superficial cells that maintain the urothelial barrier are produced via incomplete cytokinesis and endoreplication. Cell Rep., 25, 464–476.e5.
57) Pérez-Garijo, A., Martín, F.A., & Morata, G. (2004) Caspase inhibition during apoptosis causes abnormal signalling and developmental aberrations in Drosophila. Development, 131, 5591–5598.
58) Ryoo, H.D., Gorenc, T., & Steller, H. (2004) Apoptotic cells can induce compensatory cell proliferation through the JNK and the Wingless signaling pathways. Dev. Cell, 7, 491–501.
著者紹介Author Profile
 田守 洋一郎(たもり よういちろう)
田守 洋一郎(たもり よういちろう)北海道大遺伝子病制御研究所講師.理学博士.
略歴1973年大阪府生まれ.98年北海道大学理学部卒業.2000年同大学院理学研究科修士課程修了.アズワン株式会社商品開発部での2年間の勤務を経て,05年北海道大学大学院理学研究科博士課程修了(理学博士).博士号取得後渡米し,ショウジョウバエをモデルにした研究を始める.ダートマス大学で1年半,フロリダ州立大学で7年半,博士研究員として研究を行い,14年に帰国.帰国後4年間,国立遺伝学研究所にて助教として研究を続け,18年6月より現職.
研究テーマと抱負多細胞生物がどのようにして形をなし,またその形を維持するのか.その謎に魅了され,そこにある原理を理解するために研究を続けています.
ウェブサイトhttp://www.morphomeostasis.com/
趣味ギター,マウンテンバイク,料理,旅行.