不快な胃痛や胸焼けに悩まされて初めて気がつくのが,我々の胃が強い酸性環境にさらされているという事実である.重篤な場合には,胃潰瘍や逆流性食道炎と診断され,これらは直ちに生命を脅かすものではないが,患者の健康を著しく害する大変不快な症状を伴う.このような症状は,食物消化時における我々の胃が,pH 1という非常に酸性度の高い環境にさらされているために,胃酸と胃粘膜分泌のバランスが崩れることで,胃酸が自身を傷つけてしまうことに起因する.したがって,これらの疾患は胃酸分泌そのものを抑制することによって効率的に治療することができる1).一方でこの強酸性環境は,胃が担う食物(タンパク質)の消化という生理現象に本質的に重要である.胃酸は消化酵素ペプシンに至適環境を提供し,同時に食物中のタンパク質を変性させ切断を容易にする.また強酸性ゆえに細菌の繁殖を抑え,咀嚼された富栄養物を約37°Cの体温下で胃内部にとどまる2~3時間の間に腐敗させないことにも寄与している.これは,胃潰瘍発症の主原因とされているピロリ菌2)(自身でウレアーゼによってアンモニアを産生し周囲の強酸性環境を中和することができる)を例外として,病原体が体内に侵入するのを防ぐ最初のバリアとしても機能する.
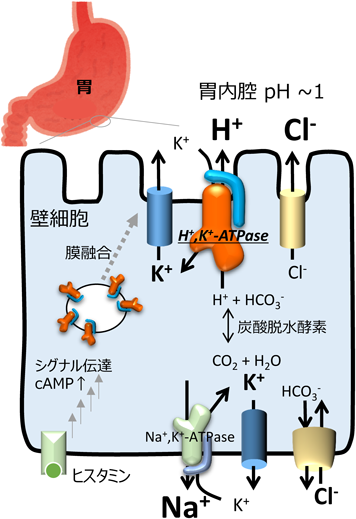
このような胃酸分泌の「酸」すなわち「H+」の輸送を担うのが,胃酸抑制剤の標的分子,胃プロトンポンプとして知られる膜タンパク質H+, K+-ATPase3, 4)である(図1).H+, K+-ATPaseはP型ATPaseファミリーに分類される膜タンパク質であり,輸送活性を担うα-subunitと補助的なβ-subunitが1:1で会合したヘテロ二量体を基本的な構成単位とする.触媒する反応はアデノシン5′-三リン酸(adenosine 5′-triphosphate:ATP)の加水分解と共役した起電性を伴わないH+とK+の対向輸送である.H+, K+-ATPaseは胃壁細胞(parietal cell)の分泌細管(secretory canaliculus)に特異的かつ大量に発現している.胃酸分泌休止時には管腔内は細胞外液から隔離されているが,細胞外からのヒスタミンによる刺激がH2受容体を介して壁細胞内でのシグナルカスケードを誘起することで,分泌細管が細胞膜と融合し,胃酸分泌すなわちH+の輸送が開始される(図1).細胞内から胃内腔へ排出される大量のH+は,静脈血由来の二酸化炭素から炭酸脱水酵素(carbonic anhydrase)によって供給される.細胞内へと過剰に流入したK+は,壁細胞に特異的かつ高い発現が認められるK+チャネル(KCNQ1–KCNE2)を介して排出される.同時にCl−がClic6をはじめとするCl−チャネルによって排出されることで,胃内部が160 mM HCl(pH 0.8)にも相当する高濃度の塩酸で満たされた状態になる5).この状況は,細胞内のpHがほぼ中性であることを考えると,細胞膜を隔ててpH差にして6ユニット,つまり100万倍のH+濃度勾配が形成されている計算になる.これほど急峻なイオン濃度勾配は,数ある能動輸送体が作り出す濃度勾配の中でも最大級といえる.このことはH+, K+-ATPaseが何らかの特殊なメカニズムを用いていることを想像させるが,それがどのようなものかは長い間明らかにされていなかった.本稿では,H+, K+-ATPaseのX線結晶構造解析と,生化学的,酵素化学的手法を用いた機能解析によって,pH 1もの強酸性溶液にH+を輸送するメカニズムの一部が理解できたので紹介する6).
P型ATPaseファミリー7)は,ATPの加水分解と共役した主としてカチオンの能動輸送を仲介する膜タンパク質として,さまざまな生命現象に密接に関わっている.このファミリーに特徴的なのは,ATPの加水分解時に末端のリン酸がタンパク質の活性中心(アスパラギン酸)に転移した自己リン酸化中間体(Phosphoenzyme intermediate:EP)を形成する反応機構であり8),これが「P」型ATPaseという呼称の由来である.このファミリーは,配列の保存性や輸送基質の特異性等からP1~P5までのサブファミリーに分類される.最もよく知られているのがP2タイプで,本稿で紹介するH+, K+-ATPaseの他にも,細胞内外にNa+とK+の電気化学ポテンシャルを形成するNa+, K+-ATPase9)や,筋肉の適切な収縮弛緩に重要なCa2+の筋小胞体への輸送をつかさどるCa2+-ATPase10)[Sarco(endo)plasmic reticulum Ca2+-ATPase 1a:SERCA]等があげられる.これらの他にも,重金属を輸送するもの(P1タイプ)や,植物のH+濃度勾配を作り出すもの(P3タイプ),イオンではなくリン脂質を基質としたフリッパーゼ活性を有するもの(P4タイプ)や輸送基質が判明していないもの(P5タイプ)まで,非常に多様なメンバーがすべての生物界において広く分布している.中でもP2タイプに属するイオンポンプの研究は古く,1957年のNa+, K+-ATPaseの発見11)以来,詳細な生化学的研究がなされ,その反応機構は生化学的によく理解されている.2000年にはP型ATPaseとして初めてSERCAのX線結晶構造12)が,2007年にはNa+, K+-ATPaseの構造13)が報告された.現在SERCAのさまざまな反応中間体の構造も含めて50以上の立体構造が報告されており,その反応機構の分子レベルでの理解が進んでいる14, 15).
P型ATPaseが行うイオン輸送反応は,Na+, K+-ATPaseに対するPost–Albers機構16, 17)に代表されるモデルによって酵素化学的に説明される.ATPの加水分解と共役した,異なる2種類のイオンの能動輸送は,酵素(「E」と省略される)がE1, E2という2種類の輸送イオンに対して異なる親和性を示すコンホメーションと,これらがそれぞれ自己リン酸化されたE1P, E2Pという,主に四状態をサイクルすることで達成される.本稿で紹介するH+, K+-ATPaseを例として説明すると(図2),H+に対して高い親和性を示すE1が細胞内側からH+を結合し,ATPを加水分解することでその末端のリン酸が活性中心に転移した自己リン酸化中間体E1Pを形成する.これがE2Pへと遷移する過程でイオン結合サイトが細胞外(胃内腔側)に向けられ,H+が細胞外へと排出される.続いてK+に高い親和性を示すE2Pが細胞外からこれを結合することで脱リン酸化され,K+を閉塞した(K+)E2中間体を形成する.閉塞されたK+は,高濃度のATP存在下,細胞内へ向けて脱閉塞,遊離する.再び細胞内側からH+が結合することによってE1中間体が形成される……という具合に,反応スキーム時計回りの方向で能動輸送サイクルが進行する4).H+, K+-ATPaseの場合,イオン輸送に起電性は認められないが(等量のカチオンが輸送される),ATP 1分子の加水分解と共役してH+とK+が一つずつ対向輸送されるという結果と,二つずつであるという結果が報告されており,そのイオン輸送化学量論はいまだ決定されていない.
これまで筆者は,ブタ胃から精製したH+, K+-ATPase標品を用いた電子線結晶学による構造解析を行い,この分子特有のメカニズムをいくつか明らかにしてきた18–24).しかしこれらの構造はヘリックスを解像する程度(6.5~8 Å)の分解能でしか得られていない.電子線結晶学は,アクアポリン25)やアセチルコリン受容体26)の原子分解能構造解析がよい例であるが,構造解析法として十分に原子分解能を達成できる.したがって問題は,H+, K+-ATPaseの二次元結晶の質に起因する.胃酸抑制剤の詳細な結合状態や,カチオンを輸送する分子機構を化学の言葉で記述するためには,原子分解能の構造が不可欠である.
一体何が,H+, K+-ATPaseの高分解能構造解析を妨げている要因なのか? 天然物(ブタ胃)からの精製は,一度に大量のタンパク質が得られるという大きな利点があるが,一方で生体内において不均一な翻訳後修飾を受けているという問題をはらんでいる.特に問題なのがβ-subunitの細胞外領域である.この部位にはブタの場合N型糖鎖付加サイトが6か所もあり,コア分子量35,000のタンパク質に対して,その倍近くの分子量の糖鎖が付加されている.さらに悪いことに,ブタ胃から精製した標品では,外から糖鎖切断酵素を加えても,その複雑な高次構造ゆえか,これらがまったく切断されない.そこで,均一な標品を得るために組換え体を利用することにした.
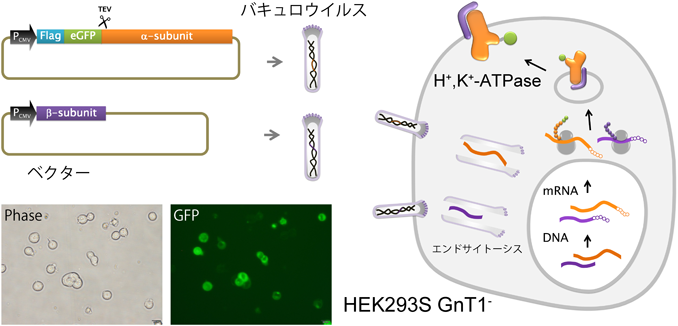
これまで多くの膜タンパク質の構造解析に実績のある昆虫細胞を用いた場合,H+, K+-ATPaseの発現量は非常に低かった.これは昆虫細胞がコレステロールを持たないこと,細胞内クォリティーコントロールの系が哺乳類と異なる等の原因が考えられた.そこで,目的遺伝子の上流にCMVプロモーターを組み込んだバキュロウイルスをベクターとして,動物細胞(HEK293)を宿主とした発現を検討したところ(BacMam,図3)27, 28),昆虫細胞と比較して40倍以上の大幅な発現量および比活性の改善がみられた.糖鎖の成熟に関わる酵素GnT1をノックアウトした細胞株を用いることで,すべてのタンパク質に付加された糖鎖がHigh-mannose型にプロセスされ,これは糖鎖切断酵素(EndoH)によって容易に除去することができる.浮遊培養による大量発現によって,今では1 L分の培養細胞から0.5 mg程度の精製標品を得ることができるようになった.この精製標品を脂質存在下で結晶化することで,多くのP型ATPaseと同様,脂質膜が積層した形の三次元結晶を得ることができた.
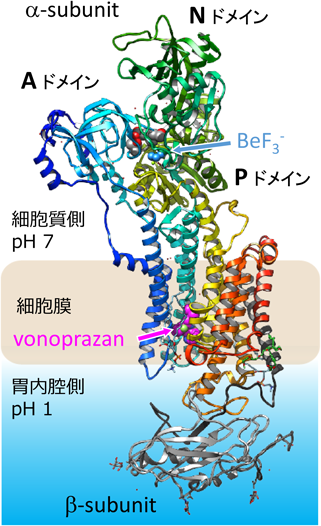
H+, K+-ATPaseの全体構造は,他のP型ATPaseと非常によく似たものであった(図4).α-subunitは細胞内にATPの加水分解に関わる三つのドメイン(A, P, Nドメイン)と,カチオン結合部位および胃酸抑制剤結合部位が存在する膜貫通領域(10本のα-helix, TM1~TM10)によって構成されている.β-subunitは1本の膜貫通ヘリックスと糖鎖付加部位の存在する細胞外ドメインで構成されている.結晶化に使用したリン酸アナログであるフッ化ベリリウム(BeF)は,Pドメインの活性中心である,P型ATPaseファミリーで普遍的に保存されたDKTGモチーフのアスパラギン酸に結合し,リン酸化中間体を模倣していた.同様に結晶化にとって必要であった胃酸抑制剤として使用されている化合物(vonoprazan)は膜貫通領域に存在する,胃内腔側(細胞外側)へ向けたくぼみに結合していた.反応機構,および他のイオンポンプの立体構造から,この構造はイオン結合サイトを胃内腔側へ向けたE2P状態に相当することがわかった.
胃酸分泌の直接の担い手であるH+, K+-ATPaseが胃酸抑制剤のターゲットとされたのは,至極当然といえよう.オメプラゾールに代表されるproton pump inhibitor(PPI)は,胃酸に関連した病態の治療に広く用いられている.かつての世界市場規模は2兆円ともいわれ,多くの胃酸に関連した病態の治療に現在も用いられている29).このPPIというクラスの薬剤は,服用時点では不活性な分子であるが,胃に到達し酸性環境によってリアクティブな化合物へと変換される.その結果,H+, K+-ATPaseに保存されたシステイン残基(Cys813)と共有結合を形成し,これを不活性化する30).胃以外に強い酸性度を持った組織は人体には存在しないので,この薬剤は胃だけで作用し,副作用の少ない,プロドラッグのお手本のような優れた薬剤である.しかしながら,いくつか改善の余地も指摘されてきた.酸性で活性化されるということは,逆にいえば酸性状態で不安定ということである.そのためPPIは腸溶剤として処方され,これが血中を巡って胃で作用するまでには一定の時間がかかる.つまり,「気持ち悪い!」と思ってPPIを服用しても,すぐには効かないわけである.また,遺伝子多型のある肝臓の薬物代謝酵素(CYP2C19)によって分解されるので,効き目に個人差があるというのも問題にされた.そこで現在では,より迅速かつ確実な治療を目指して,新しいクラスの薬剤が開発された.2015年にわが国で上市されたvonoprazan(商品名:タケキャブ,武田薬品工業)は,その先駆けであり,胃プロトンポンプを直接ブロックすることで,迅速な治癒を実現した31).これら新しいクラスの薬剤は,H+, K+-ATPase活性をK+競合的に阻害することから,P-CABs(potassium-competitive acid blockers)と呼ばれる.
これらの薬剤は,胃酸過多の治療の他にもピロリ菌除菌の際の補助薬として用いられている.ピロリ菌は胃がんの発症との関連が指摘されており,保菌者には基本的に除菌が推奨される.除菌は2種類の抗生物質を同時に服用することで,その相乗効果により高い除菌率が達成されるが,このとき抗生物質が胃酸によって分解されてしまうのを防ぐために,胃酸抑制剤が併用される.
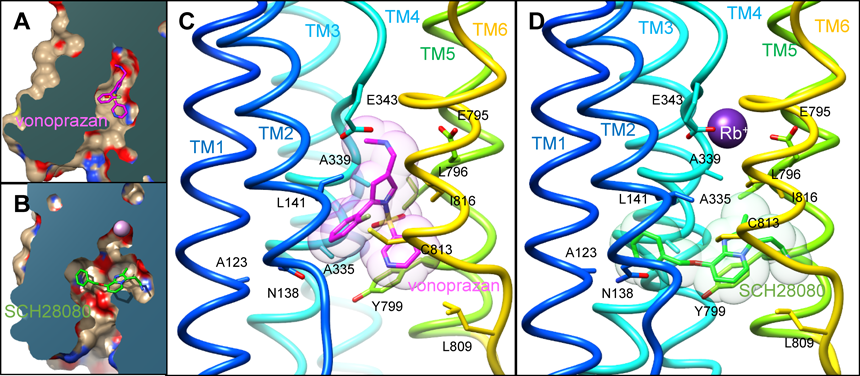
H+, K+-ATPaseの結晶構造は,vonoprazan31)とSCH2808032)という,2種類の化合物との複合体として得られた(図5).実際に臨床で用いられているvonoprazanは,胃内腔側からカチオン結合サイトをつなぐ,いわば「通路」の中ほどに,これを塞ぐように結合していた(図5A).こうなってしまっては対向輸送イオンであるK+がカチオン結合サイトにたどり着くのは難しい.文字どおり,K+の結合を「ブロック」しているわけである.これが,vonoprazanがK+競合的な阻害作用を示す構造的な理由である.一方SCH28080は,その細胞毒性ゆえに薬剤としては利用されていないが,現在開発中のいくつかのP-CABに基本骨格を提供するプロトタイプ化合物として,以前からin vitroにおける特異的阻害剤として利用されてきた.SCH28080も,同様にカチオンの通路を塞ぐ形で結合している(図5B).しかしながら,化合物はカチオン結合サイトの手前に結合しており,カチオン結合サイトにはK+の同族体であるRb+(K+と同様の作用を示す)が結合していた.このことから,P-CABの示すK+競合的な阻害作用は,実際にK+結合サイトを奪い合っているわけではなく,P-CABの結合によってK+閉塞構造への遷移が妨げられることに起因することが明らかになった.
原子モデルによって,いまやどのアミノ酸がそれぞれの化合物の結合に重要なのかが理解できる(図5C, D, 表1).Try799はvonoprazanおよびSCH28080どちらの化合物の結合に対しても重要であり,結合サイトにフタをするような形で化合物の解離を防ぎ,それぞれの高い親和性に大きく関与する.Tyr799Phe変異体ではフタとしての効果が十分発揮されるのでそれぞれの化合物に対する親和性に大きな影響はないが,Tyr799Alaのように小さいアミノ酸へと置換することによって,両者の薬剤に対して著しい結合親和性の低下がみられる(表1).これとは逆に,たとえばTM1に存在するAla123はSCH28080のベンゼン環に近い位置に存在するが,vonoprazanの結合部位からはかなり離れたところにある.したがって,このアミノ酸を大きなものに変異させると,SCH28080の結合親和性にのみ強い影響がみられる.部位特異的変異体のデータは,これらの化合物の結合構造をよく説明するものであった.また,これら結合に関与するアミノ酸は,疎水的なものがほとんどであることが特徴である(図5C, D, 表1).この疎水的な結合ポケットに隙間なくはまり込むことで(図5A, B),化合物の周囲に存在していた水和水が排除され,系全体に好ましいエントロピーの上昇がもたらされる.このような疎水的な相互作用によって薬剤が結合することは,たとえば静電的な相互作用よりも,胃の表面で働く上では理想的であるといえる.なぜなら,胃の中のpH 1という環境では,ほとんどのカルボキシ基にH+が結合して電荷を持たない状態になっているので,たとえば静電的な相互作用を狙った薬剤よりも,疎水的な相互作用を主とする薬剤の方が結合に有利であると考えられるからである.
表1 変異体の胃酸抑制剤に対する親和性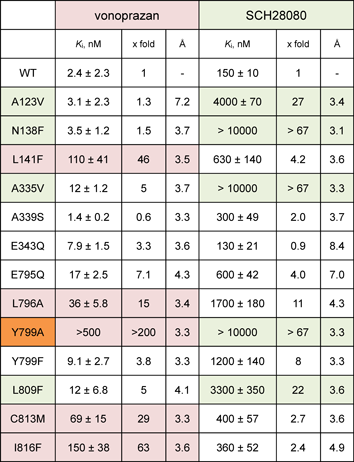 |
| 各阻害剤に対する阻害定数(Ki)の変移導入による変化.野生型のKiと比較して何倍悪くなったか(-fold)と,結晶構造中における化合物とタンパク質間の最短距離(Å)を合わせて記した. |
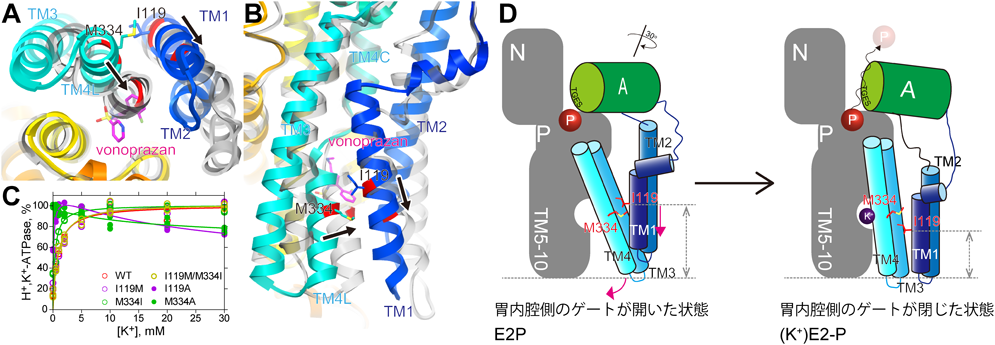
他のP型ATPaseや,H+, K+-ATPaseの電子線結晶学による構造解析から,胃内腔(細胞外)への輸送イオンのゲーティングの際に引き起こされる構造変化はよく理解されている21–23, 33, 34).胃内腔へ開いたゲートから対向輸送イオンであるK+が侵入,カチオンサイトに結合することに端を発し,TM1-2およびTM3-4のヘリックスが動くことでゲートは閉じられる.TM1-2はAドメインへとつながっており,TMの構造変化が細胞内のAドメインに伝わることで,このドメインがPドメインに対して相対的に回転し,Pドメインに結合したリン酸が加水分解される.このようなゲートを閉じる一連のアロステリックな過程において鍵となるのは,TM1-2ヘリックスの上下運動である.ゲートが開いているときに,TM1-2は細胞質側に存在しているが,ゲートが閉じるときには胃内腔側へとスライドする(図6D).今回構造解析された,胃内腔側のゲートが開いた状態である薬剤結合構造では,TM1のIle119が,ちょうど留め金(latch)のように,TM4に存在するMet334と折り重なるようにして存在し,TM1-2ヘリックスが胃内腔側へとスライドするのを防いでいるようにみえた(図6A, B).もしそうであれば,これらの側鎖のうちのどちらかをより小さなAlaに置換することで,TM1-2ヘリックスが細胞質側にとどまることができずに胃内腔側へと自発的にスライドし,細胞外側へのゲートは閉じてしまうはずである.ゲートが閉じると,上述のとおりそのシグナルはAドメインへと伝播し,最終的には脱リン酸化が誘起される.野生型においてこの脱リン酸化反応は,K+が結合することによって引き起こされるわけであり(図2, E2P→K+E2),したがって野生型の場合,ATPase活性はK+非存在下では非常に低く,K+濃度に依存して上昇していく(図6C).これとは対照的に,Met334AlaやIle119Ala変異体ではK+濃度に依存しないconstitutive-activeなATPase活性が観測された(図6C).一方で,Ile119MetやMet334Ileおよび双方を入れ替えた変異体では,野生型と同様のK+に依存したATPase活性がみられた.これらの結果は,Ile119とMert334の疎水的な相互作用が,TM1-2ヘリックスの胃内腔側へのスライドを防ぐ“latch”のような機能を果たすことによって,ゲーティングと脱リン酸化の共役に重要な役割を担っていることを示している(図6D).したがって,上記のような相互作用が観察された今回の立体構造は,決して薬剤の結合によって誘発された人工的なコンホメーションではなく,多くのタンパク質の薬剤結合構造がそうであるように,薬剤の結合がそのタンパク質が生体内でとりうる一状態に結合しこれを安定化することで得られた,胃内腔側へのゲートが開いた状態を捉えていると考えられる.
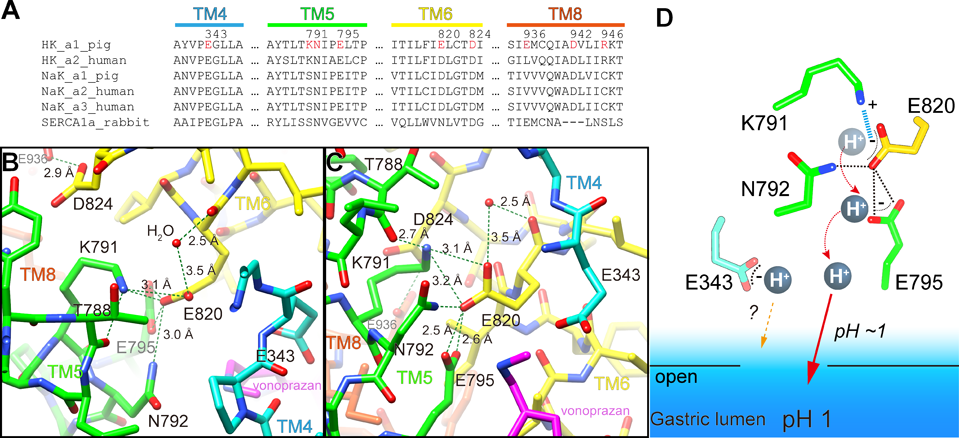
反応機構に従えば,E2P状態は,H+を輸送した直後,次にK+が入ってくる直前の状態である(図2).したがって,この状態におけるカチオン結合サイトを見れば,H+がどのように排出されるのかが理解できる.他のP型ATPaseやカチオン輸送体と同様,H+, K+-ATPaseも膜貫通領域にある酸性アミノ酸をH+の結合・解離に用いていると考えられる.これら酸性アミノ酸は,H+, K+-ATPaseにおいても,近縁のNa+, K+-ATPaseにおいてもよく保存されている(図7A).H+, K+-ATPaseの場合,三つのグルタミン酸がカチオン結合サイトに存在する.しかしながら,一般的な酸性側鎖の酸解離定数(pKa)は,3~4程度である.この値は,これら酸性側鎖からH+を解離できるのはせいぜいpH 3~4の溶液までで,pH 1の溶液に対してはほとんどH+を遊離しない,ということを意味している.しかし,我々の胃の中にはpH 1の溶液が間違いなく存在している.我々が摂取したタンパク質を消化し,飲み過ぎた次の日に胸焼けを感じるのが何よりの証左であろう.したがって,H+, K+-ATPaseは何か特殊な方法で,H+を胃の強酸性溶液に送り出しているはずである.
カチオン結合サイトの構造(図7B, C)で,まず目を引くのが二つのグルタミン酸の近接構造である.Glu820はGlu795と実に2.5 Åという非常に近い距離にある.負に帯電しているグルタミン酸どうしが,これほどの近接構造をとることはありえないので,この近接構造は少なくともどちらか片方のグルタミン酸がH+を結合して電気的に中性であることを意味している.Glu795Gln変異体が野生型とほとんど変わらない活性プロファイルを示すことから6, 35, 36),この場合Glu795がプロトネーションされていると考えられた.このような酸性側鎖の近接構造は,多くの酵素の活性中心にみられる.同じように胃で働くペプシンもその一つである.ペプシンの場合,活性中心における二つのアスパラギン酸が2.5 Åの距離にある.この近接構造により,互いのpKaがそれぞれ1.4と4.7という値になり,これがペプチド結合の加水分解の鍵となる37, 38).H+, K+-ATPaseにおけるGlu795とGlu820の近接構造でも,おそらく似たような状況が生じている.つまり,Glu795のpKaが高くなり(結果H+を結合),逆にGlu820のpKaは低下し,H+を解離しやすくなる.
しかし,おそらくこれだけではpH 1の水溶液に対してH+を放出するには不十分だったのであろう.カチオン結合サイトには,もう一つ重要な相互作用がみられた.Lys791はカチオン結合サイトに存在する唯一の塩基性アミノ酸である.このリシン残基はH+, K+-ATPaseにおいて普遍的に保存されているが,Na+, K+-ATPaseやSERCAではセリンに置換されている(図7A).カチオンを結合する場所にあって,塩基性アミノ酸の果たす役割とは何であるのか? H+, K+-ATPaseの一次配列が決定されたときから,このリシンの役割は問題にされてきた39–42).結晶構造をみると,Lys791は,Glu820と近接した位置(3.1 Å)にあり,これと静電的な相互作用をしていることがわかった.このプラス-マイナスの相互作用によって,Glu820はもはやH+を保持していることができなくなり,たとえそれがpH 1の強酸性水溶液であっても,強制的にカチオン結合サイトから排出される.Glu820は溶液に面していないので,Glu795との水素結合ネットワークを介して,たとえるならビリヤードの玉のようにして,H+が排出される(図7D).このような仕組みによって,H+, K+-ATPaseが胃の中に強い酸性環境を作り出していることが理解できた.
本稿で紹介した構造-機能解析に基づいて,なぜH+, K+-ATPaseが胃内部にpH 1という環境を実現できるかという問題が一部説明できるようになった.しかしながら,今回紹介した構造はH+を排出した“直後”の構造である.したがって多くの疑問がいまだのこされたままであることを最後に記しておきたい.酸性に向かってH+を押し出すことは理解できたが,逆にH+濃度の低い(中性)溶液からどのようにH+を汲み上げるのか? Lys791によるH+の押し出しは何によって駆動されるのか? 対向輸送されるK+はどのように結合して反応サイクルを進めるのか? イオン輸送化学量論は43, 44)?E343からH+は離れるのか?等々である.このような新たな問いに答えるためには,ダイナミックに構造変化するプロトンポンプの反応機構を分子ムービーとして理解する必要があり,個々の反応中間体の構造解析が急務である.
謝辞Acknowledgments
本稿で紹介したH+, K+-ATPaseの構造機能解析に関して,名古屋大学細胞生理学研究センター 藤吉好則,入江克雅,中西華代,鈴木博視(現Rockefeller Univ.)各博士のご尽力に感謝致します.北海道大学の谷口和弥博士(故人)には,プロジェクトの発足時から通して数多くの有用な助言を賜りましたことに感謝致します.
引用文献References
1) Sachs, G., Shin, J.M., Vagin, O., Lambrecht, N., Yakubov, I., & Munson, K. (2007) The gastric H, K ATPase as a drug target: past, present, and future. J. Clin. Gastroenterol., 41(Suppl 2), S226–S242.
2) Marshall, B.J. & Warren, J.R. (1984). Lancet, 1, 1383–1401.
3) Ganser, A.L. & Forte, J.G. (1973) K+-stimulated ATPase in purified microsomes of bullfrog oxyntic cells. Biochim. Biophys. Acta, 307, 169–180.
4) Rabon, E.C. & Reuben, M.A. (1990) The mechanism and structure of the gastric H, K-ATPase. Annu. Rev. Physiol., 52, 321–344.
5) Shin, J.M., Munson, K., & Sachs, G. (2009) The gastric HK-ATPase: structure, function, and inhibition. Eur. J. Phys., 457, 609–622.
6) Abe, K., Irie, K., Nakanishi, H., Suzuki, H., & Fujiyoshi, Y. (2018) Crystal structures of the gastric proton pump. Nature, 556, 214–218.
7) Palmgren, M.G. & Axelsen, K.B. (1998) Evolution of P-type ATPases. Biochim. Biophys. Acta, 1365, 37–45.
8) Post, R.L. & Kume, S. (1973) Evidence for an aspartyl phosphate residue at the active site of sodium and potassium ion transport adenosine triphosphatase. J. Biol. Chem., 248, 6993–7000.
9) Jørgensen, P.L., Hákansson, K.O., & Karlish, S.J.D. (2003) Structure and mechanism of Na, K-ATPase:functional sites and their interactions. Annu. Rev. Physiol., 65, 817–849.
10) Møller, J.V., Olesen, C., Winther, A.-M.L., & Nissen, P. (2010) The sarcoplasmic Ca2+-ATPase: design of a perfect chemi-osmotic pump. Q. Rev. Biophys., 43, 501–566.
11) Skou, J.C. (1957) The influence of some cations on an adenosine triphosphatase from peripheral nerves. 1957. Biochim. Biophys. Acta, 1000, 439–446.
12) Toyoshima, C., Nakasako, M., Nomura, H., & Ogawa, H. (2000) Crystal structure of the calcium pump of sarcoplasmic reticulum at 2.6 A resolution. Nature, 405, 647–655.
13) Morth, J.P., Pedersen, B.P., Toustrup-Jensen, M.S., Sørensen, T.L., Petersen, J., Andersen, J.P., Vilsen, B., & Nissen, P. (2007) Crystal structure of the sodium–potassium pump. Nature, 450, 1043–1049.
14) Toyoshima, C. (2008) Structural aspects of ion pumping by Ca2+-ATPase of sarcoplasmic reticulum. Arch. Biochem. Biophys., 476, 3–11.
15) Toyoshima, C. & Cornelius, F. (2013) New crystal structures of PII-type ATPases:excitement continues. Curr. Opin. Struct. Biol., 23, 507–514.
16) Post, R.L., Hegyvary, C., & Kume, S. (1972) Activation by adenosine triphosphate in the phosphorylation kinetics of sodium and potassium ion transport adenosine triphosphatase. J. Biol. Chem., 247, 6530–6540.
17) Albers, R.W. (1967) Biochemical aspects of active transport. Annu. Rev. Biochem., 36, 727–756.
18) Abe, K., Tani, K., Nishizawa, T., & Fujiyoshi, Y. (2009) Inter-subunit interaction of gastric H+, K+-ATPase prevents reverse reaction of the transport cycle. EMBO J., 28, 1637–1643.
19) Dürr, K., Abe, K., Tavraz, N.N., & Friedrich, T. (2009) E2P state stabilization by the N-terminal tail of the H, K-ATPase beta-subunit is critical for efficient proton pumping under in vivo conditions. J. Biol. Chem., 284, 20147–20154.
20) Abe, K., Tani, K., & Fujiyoshi, Y. (2010) Structural and functional characterization of H+, K+-ATPase with bound fluorinated phosphate analogs. J. Struct. Biol., 170, 60–68.
21) Abe, K., Tani, K., & Fujiyoshi, F. (2011) Conformational rearrangement of gastric H+, K+-ATPase induced by an acid suppressant. Nat. Commun., 2, 155.
22) Abe, K., Tani, K., Friedrich, T., & Fujiyoshi, Y. (2012) Cryo-EM structure of gastric H+, K+-ATPase with a single occupied cation-binding site. Proc. Natl. Acad. Sci. USA, 109, 18401–18406.
23) Abe, K., Tani, K., & Fujiyoshi, Y. (2014) Systematic comparison of molecular conformations of H+, K+-ATPase reveals an important contribution of the A-M2 linker for the luminal gating. J. Biol. Chem., 289, 30590–30601.
24) Abe, K., Shimokawa, J., Naito, M., Munson, K., Vagin, O., Sachs, G., Suzuki, H., Tani, K., & Fujiyoshi, Y. (2017) The cryo-EM structure of gastric H+, K+-ATPase with bound BYK99, a high-affinity member of K+-competitive, imidazo[1,2-a]pyridine inhibitors. Sci. Rep., 7, 6632.
25) Murata, K., Mitsuoka, K., Hirai, T., Walz, T., Agre, P., Heymann, J.B., Engel, A., & Fujiyoshi, Y. (2000) a. Nature, 407, 599–605.
26) Miyazawa, A., Fujiyoshi, Y., & Unwin, N. (2003) a. Nature, 423, 949–955.
27) Dukkipati, A., Park, H.H., Waghray, D., Fischer, S., & Garcia, K.C. (2008) a. Protein Expr. Purif., 62, 160–170.
28) Goehring, A., Mitsuoka, K., Hirai, T., Walz, T., Agre, P., Heymann, J.B., Engel, A., & Fujiyoshi, Y. (2014) Structural determinants of water permeation through aquaporin-1. Nat. Protoc., 9, 25742585.
29) Shin, J.M. & Sachs, G. (2008) Pharmacology of proton pump inhibitors. Curr. Gastroenterol. Rep., 10, 528–534.
30) Shin, J.M., Cho, Y.M., & Sachs, G. (2004) Chemistry of covalent inhibition of the gastric (H+, K+)-ATPase by proton pump inhibitors. J. Am. Chem. Soc., 126, 7800–7811.
31) Arikawa, Y., Hasuoka, A., Nishida, H., Hirase, K., Inatomi, N., Takagi, T., Tarui, N., Kawamoto, M., Imanishi, A., Itoh, F., et al. (2015) Synthetic studies of five-membered heteroaromatic derivatives as potassium-competitive acid blockers (P-CABs). Bioorg. Med. Chem. Lett., 25, 2037–2040.
32) Kaminski, J.J., Bristol, J.A., Puchalski, C., Lovey, R.G., Elliott, A.J., Guzik, H., Solomon, D.M., Conn, D.J., Domalski, M.S., Wong, S.C., et al. (1985) Antiulcer agents. 1. Gastric antisecretory and cytoprotective properties of substituted imidazo[1,2-a]pyridines. J. Med. Chem., 28, 876–892.
33) Toyoshima, C., Norimatsu, Y., Iwasawa, S., Tsuda, T., & Ogawa, H. (2007) How processing of aspartylphosphate is coupled to lumenal gating of the ion pathway in the calcium pump. Proc. Natl. Acad. Sci. USA, 104, 19831–19836.
34) Danko, S., Yamasaki, K., Daiho, T., & Suzuki, H. (2004) Distinct natures of beryllium fluoride-bound, aluminum fluoride-bound, and magnesium fluoride-bound stable analogues of an ADP-insensitive phosphoenzyme intermediate of sarcoplasmic reticulum Ca2+-ATPase: changes in catalytic and transport sites during phosphoenzyme hydrolysis. J. Biol. Chem., 279, 14991–14998.
35) Swarts, H.G.P., Hermsen, H.P.H., Koenderink, J.B., Stekhoven, F.M.A.H.S., & De Pont, J.J.H.H.M. (1998) A conformation-specific interhelical salt bridge in the K+ binding site of gastric H, K-ATPase. EMBO J., 17, 3029–3035.
36) Swarts, H.G.P., Koenderink, J.B., Hermsen, H.P.H., Willems, P.H.G.M., & De Pont, J.J.H.H.M. (2001) K+-independent gastric H+, K+-atpase activity. Dissociation of K+-independent dephosphorylation and preference for the E1 conformation by combined mutagenesis of transmembrane glutamate residues. J. Biol. Chem., 276, 36909–36916.
37) Sielecki, A.R., Fedorov, A.A., Boodhoo, A., Andreeva, N.S., & James, M.N.G. (1990) Molecular and crystal structures of monoclinic porcine pepsin refined at 1.8 A resolution. J. Mol. Biol., 214, 143–170.
38) Clement, G.E., Rooney, J., Zakheim, D., & Eastman, J. (1970) The pH dependence of the dephosphorylated pepsin-catalyzed hydrolysis of N-acetyl-L-phenylalanyl-L-tyrosine methyl ester. J. Am. Chem. Soc., 92, 186–189.
39) Koenderink, J.B., Swarts, H.G.P., Willems, P.H.G., Krieger, E., & De Pont, J.J.H.H.M.A. (2004) A conformation-specific interhelical salt bridge in the K+ binding site of gastric H, K-ATPase. J. Biol. Chem., 279, 16417–16424.
40) Munson, K., Garcia, R., & Sachs, G. (2005) Inhibitor and ion binding sites on the gastric H, K-ATPase. Biochemistry, 44, 5267–5284.
41) Burnay, M., Crambert, G., Kharoubi-Hess, S., Geering, K., & Horisberger, J.D. (2003) Electrogenicity of Na, K- and H, K-ATPase activity and presence of a positively charged amino acid in the fifth transmembrane segment. J. Biol. Chem., 278, 19237–19244.
42) Dürr, K., Seuffert, I., & Friedrich, T. (2010) Deceleration of the E1P-E2P transition and ion transport by mutation of potentially salt bridge-forming residues Lys-791 and Glu-820 in gastric H+/K+-ATPase. J. Biol. Chem., 285, 39366–39379.
43) Reenstra, W.W. & Forte, J.G. (1981) H+/ATP stoichiometry for the gastric (K+ + H+)-ATPas. J. Membr. Biol., 61, 55–60.
44) Rabon, E.C., McFall, T.L., & Sachs, G. (1982) The gastric [H, K]ATPase: H+/ATP stoichiometry. J. Biol. Chem., 257, 6296–6299.
著者紹介Author Profile
 阿部 一啓(あべ かずひろ)
阿部 一啓(あべ かずひろ)名古屋大学細胞生理学研究センター・大学院創薬科学研究科准教授.博士(理学).
略歴1975年北海道に生る.2004年北海道大学大学院理学研究科化学専攻博士後期課程修了,京都大学生物物理学教室を経て,15年より現職.
研究テーマと抱負能動輸送体の生化学,構造生理学.
ウェブサイトhttp://www.cespi.nagoya-u.ac.jp/
趣味バスケットボール,ソフトボール,ビール,ウイスキー.