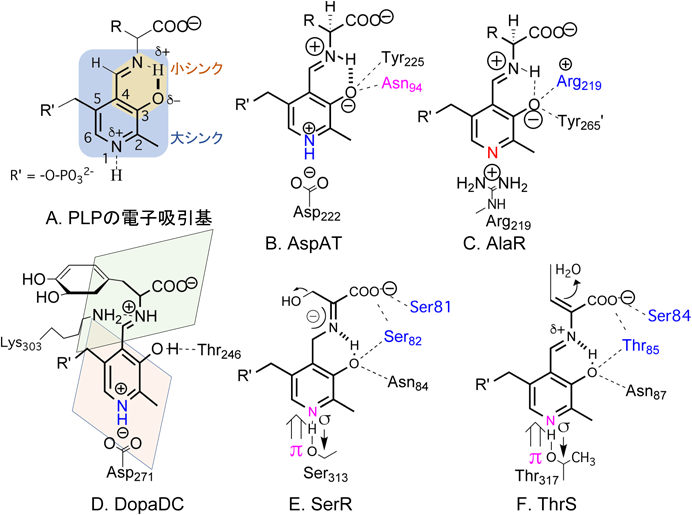
1. 一つの補酵素から多様な反応を引き出すPLP酵素
酵素反応は多段階の素反応の積み重ねで進行しており,各ステップを理解するには遷移状態や中間体の構造とエネルギーを解析する必要がある.X線結晶構造解析に量子化学計算を導入すれば基質や中間体へのプロトン付加状態,遷移状態などの動的な化学構造が予測できる1, 2).つまりアミノ酸残基や補酵素の化学構造を原子のレベルから電子のレベルに掘り下げて,化学反応がどのように進行するかという考察を構築することが可能になる.
タンパク質を構成するアミノ酸残基はpKa 3~16に及ぶ多様な解離基を持っているが,水和状態ではすべて求核剤として働く.酵素の約7割が活性中心に補酵素または金属を持つのは,補因子の多くが求電子剤としての機能を発揮するためである.ピリドキサール5′-リン酸(pyridoxal phosphate:PLP)もまた,電子求引性を発揮してアミノ基転移,脱炭酸,ラセミ化,α, β位またはγ位の共有結合を切断または形成する反応に関わる.これら多彩な反応における補酵素PLPの役割については,林3),吉村4)による優れた総説があり,PLP酵素の反応機構が包括的に紹介されている.補酵素PLPからどの反応を引き出すのかを決定するのは,補酵素の周辺に配置されたアミノ酸残基群である.PLPを取り囲む残基群は,PLPのプロトンの付加状態とそのエネルギー,反応経路における遷移状態構造を支配しており,多様な反応性は多様な酵素・補酵素の相互作用によって発揮されているといえる.
基質とシッフ塩基を介して複合体を構成した補酵素PLPは,アルジミン中間体と呼ばれる.このアルジミン構造には,電子を引き込むシンクが大小二つあると有機電子論的には解釈されてきた.大きなシンクは正電荷を帯びたピリジン環とシッフ塩基を含む広い共役系を持つ(図1A).一方,小シンクとは分子内水素結合を持つシッフ塩基のみで,吸引された電子はピリジン環Nまでは降りてこない.大シンクを活用する代表的PLP酵素はアミノ基転移酵素であり,ピリジン環に配位したAsp残基が正電荷を保持している(図1B).小シンクを活用するPLP酵素の代表はアミノ酸ラセマーゼで,グアニジド基に正電荷を持つArg残基を配置してピリジン環Nには正電荷を持たせない形をとる(図1C).脱炭酸酵素は少し手が込んでいて,ピリジン環にAsp残基を配置しながらもピリジン環平面とシッフ塩基の平面を直交させることで二つの平面上のπ電子系が同一平面で共役できない構造をあえてとる(図1D).アラニンラセマーゼ(AlaR)とドーパ脱炭酸酵素(DopaDC)にみられる小シンク活用型PLPの求電子性を理解する上で,注目すべきは3位のO3フェノール基である.周囲のアミノ酸残基からO3にプロトンが与えられると,小シンクのイミド基側にプロトンが押しつけられて正荷電が強められるので電子吸引性は高くなる(図1C).逆にO3が解離してフェノラート化する作用があれば,分子内水素結合にあずかるプロトンがシッフ塩基部分から離れてO3に引っ張られるためにイミド基の電子吸引性は弱められる.σ結合やπ電子の出入りには,大小のシンクの活用と解離状態の制御によって発揮されている精緻なメカニズムが存在する.
2. アスパラギン酸アミノ基転移酵素(AspAT)
PLPが基質と結合したアルジミン構造はPLP酵素反応に共通する中間体であり,ここを分岐点として多様な触媒反応が導かれてゆく.つまり,PLP酵素の反応の多様性は,この構造のどの電子がPLP内のどこまで吸引されるかで決定される.大腸菌のアスパラギン酸アミノ基転移酵素(AspAT)では,基質のα水素が引き抜かれて生じる負電荷(カルボアニオン)がPLPのピリジンNまでの広い共役系に引き込まれて安定なキノノイド中間体を形成する(図2A).大シンクへの電子吸引によってカルボアニオンは安定化する.続いて引き抜かれたプロトンがC4位に付加されてケト酸が遊離し,ピリドキサミンリン酸(pyridoxamine phosphate:PMP)が残される.酵素反応を理解する上で鍵となるのは,PLP周辺のアミノ酸残基群がキノノイド型に誘導する仕組みである.
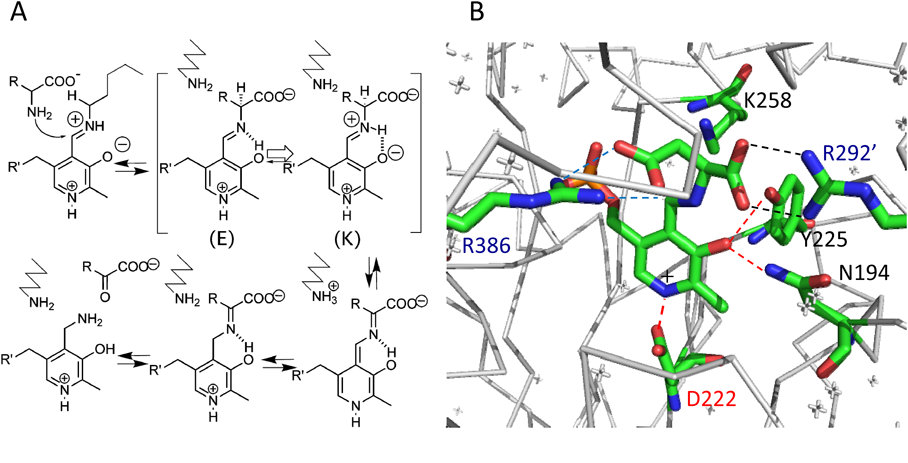
Chandraらは,比較的高精度な量子化学計算を実施できる基底関数B3LYP/6-311G**:PM3を用いてAspATの外部アルジミン状態におけるケトエナミン(K)型とエノールイミン(E)型の互変異性反応を計算した5).この計算では,外部アルジミン状態Asp–PLPと周辺の6アミノ酸残基が計算対象となった.周辺6残基とは,基質Aspのα, β-カルボキシル基と相互作用するArg386, Arg292′の両残基,ピリジン環Nにプロトン供与するAsp222, PLPのフェノラートO3に相互作用するAsn194とTyr225,さらに基質が入るまでPLPと結合していたLys258である.各残基のβ炭素より先が量子化学計算の対象とされており,α炭素を含む主鎖構造は量子化学計算の対象外となっている(図2B).
これらの残基のうちPLPに作用するのはピリジンNへ正電荷を付与するAsp222であり,解離したO3のフェノラートアニオンに対してはAsn94のアミド水素とTyr225のOH(水酸基)が安定化に寄与している.活性中心においてK型はE型よりも7 kcal/mol安定で,K型からE型へのエネルギー障壁は7.5 kcal/molと算出された.AspATの活性中心においてAsp–PLP複合体はK型の電子状態に誘導されており,α水素が引き抜かれてカルボアニオンが生じると速やかに大シンクまで電子対が移動してエネルギー的にも安定化することが示された.
Geobacillus stearothermophilusのアラニンラセマーゼ(AlaR)ではピリジン環Nに正電荷を持つArg残基が配位しており,ピリジン環Nは正荷電を帯びない状態にある(図1C).AlaRの場合も外部アルジミン構造からα水素が引き抜かれて負電荷が生成するが,カルボアニオンがピリジンNまで拡がった状態は不安定なのでα炭素へのプロトン付加による立体反転が進行してラセマーゼとして働く.大シンクへの電子吸引がエネルギー的に不利になる機構を電子レベルで解明するために,AlaRの量子化学計算は基質AlaとPLPとの外部アルジミン構造,およびそれを取り巻く7残基で行われた.
ラセマーゼ反応は二塩基機構であり,Ala–PLP複合体からα水素を引き抜く残基として,L-Ala–PLPに対してはTyr265′のフェノラートアニオン−O−が,D-Ala–PLPの場合はLys39がそれぞれ働く.さらにAlaRがAspATと異なる点は,ピリジン環のO3の近傍残基に強い正電荷を持つArg136残基が配置されていることであり,O3の負電荷を保つ効果がある.これによりシッフ塩基に正荷電が誘導されるので,大シンクの働きは抑制しつつ,小シンクの電子求引性を高める役割を担う.大シンクほどの強い電子吸引性がないことは,ラセマーゼには好都合といえる.ハミルトニアンAM1を用いた半経験的分子軌道計算によってシミュレーションした反応予測では,活性化エネルギーや速度論解析データともよく一致した6).ハミルトニアンAM1はプロトン引き抜きのような酸塩基反応では実験値とよく一致することが経験的に知られているので,プロトンを授受するような酸塩基反応の解析には有用といえる.
Majorらは,AlaRのPLPのピリジン環Nに酸性残基を配置したR219E型酵素と野生型酵素についても反応経路の計算シミュレーションを実施した.αプロトンが引き抜かれて生成したカルボアニオンが非局在化してピリジン環にまで拡大すると,より安定なキノノイド状態に陥ることが予測され,アミノ基転移の半反応が進行する過程も計算によって示された.
PLPから大・小シンクの電子状態がどのように引き出されるのかを理解する上で,AspATとAlaRは対照的である.これらのPLP酵素の仕組みを理解することは,ドーパ脱炭酸酵素とセリンラセマーゼ/脱水酵素の反応機構の理解にも役立つ.
ドーパ脱炭酸酵素(DopaDC)はAspATと同様にピリジン環Nに酸性残基Asp271を配しており,ピリジン環に強い電子吸引性を付与する点では大シンク活用型と同じである.しかし,DopaDC酵素の活性中心に置かれた外部アルジミン構造を量子化学で高精度に計算するとAspATの場合とは異なり,ややE型優位(−1.5 kcal/mol)な電子状態をとると計算された7).実験によればDopaDCは外部アルジミン構造が特異な吸収極大(380 nm)を持つことからも酵素内部での平衡はE型優勢であることが支持されており,さらにシッフ塩基の平面とピリジン環が同一平面ではなく,二つの面が直交している可能性が指摘されてきた8).その先見的な考察は,量子計算化学からも強く支持されており,このE型の外部アルジミン構造ではシッフ塩基の平面とピリジン環が79.8±34.7°の大きな二面角でほぼ直交した構造として保持されており,そのような二面角が触媒作用にも必須と考えられた(図1D).周辺のアミノ酸残基の作用によりPLP分子内のイミン窒素Nとフェノール基O3はプロトンを共有できず,引き離された状態をとる.つまりThr246とフェノール基O3の間に水素結合が作られ,イミンNにはLys303残基が3 Åの近傍から水素結合を提供している.これらの周辺アミノ酸残基群によってイミンNとフェノール基O3は,同一平面で水素結合を作らず,二つの面が直交した基質−PLPという特異な構造を作っている.
DopaDC反応について反応シミュレーションを行った結果,E型からの脱炭酸がより低い自由エネルギー経路をたどって脱炭酸反応を完結することが示されたので,E型状態に合理性があると考えられた.この経路積分計算においては,切れる結合と形成される結合にあらかじめ自由度を高くした設定で計算コストを節約する計算方法がXieらによって開発されており9),Tonyらはこれを用いて経路積分の計算コストを節約している7).
フェノールが非解離状態に維持されて二つの面が直交することでシッフ塩基の平面とピリジン環は共役せず,カルボアニオン電子をピリジン環に流入させない.結果的にキノノイド中間体を形成せずα炭素に速やかにプロトンを受け取ることで脱炭酸の素反応が完結する.この反応過程も計算機上でシミュレーションされた.不可逆的に進行する脱炭酸過程では結晶構造解析で観測できる中間体が形成されないので反応機構に関わる知見が乏しい.量子化学計算を導入することで周辺残基とPLPの相互作用を電子のレベルで解明する考察が提供されたといえる.
5. セリンラセマーゼ/脱水酵素(SerR/DH)とトレオニン合成酵素(ThrS)
SerやThrのβ位水酸基からの脱水反応,または水酸基の付加を触媒するPLP酵素群は,ピリジンNにSer残基またはThr残基を配している.ピリジンNに明白な正電荷は発生しないがN…H水素結合距離は短く,正電荷を持つピリジン環と同様にカルボアニオン中間体の安定化に寄与すると考えられる.なぜ,これらの酵素群ではSerまたはThr残基がPLPのNに配位するのか,という疑問を念頭において行った量子化学計算の取り組みを2件紹介したい.
ラットのセリンラセマーゼ(SerR)は,脳内でNMDA受容体の活性化に関わるD-セリンをL体から生成するラセミ化反応と,セリンの脱水反応による分解反応を触媒する.SerRによるラセミ化反応も二塩基機構であり,基質PLPの平面構造をはさんで二つの塩基(Lys56とSer84)が酸塩基触媒として働いてラセミ化反応が進行する.L-セリンが基質のときLys56残基がα水素を奪い,D-セリンが基質のときSer84残基が塩基として作用する.LysとSerでは塩基性に大きな差があるが,Majorらによる量子化学計算による活性化エネギーは同等の値が算出された.いずれの基質からα水素が引き抜かれても同じカルボアニオン中間体が形成される(図1E).SerRの反応機構においては基質α炭素に発生するカルボアニオンを安定化する仕組みが重要で,基質のカルボキシル基を固定する特定の残基は存在せず,Ser81-Ser82-X-Asn84の主鎖のアミド基が提供する正電荷群がオキシアニオンホールによって安定化される.
Majorらは,AlaRで確立した半経験的分子軌道計算を用いてSerRによるL→D,D→Lのラセミ化反応に伴う自由エネルギー変化をプロットした.さらに量子化学計算による結果でもAM1による計算とほぼ一致する反応経路が描かれた.L-Serを基質としてLys56がプロトンを引き抜く反応では17.8 kcal/molの活性化エネルギー,D-Serが基質となる反応では20.7 kcal/molの活性化エネルギーを要する.後者がやや高いのは,Ser84残基が負電荷を持つ状態に活性化するために3 kcal/molのエネルギーを要するためと見積もられた.
小シンクの電子吸引性を調節するのがPLPのフェノラート基O3の解離状態で,これに作用するAsn84残基は本酵素には高度に保存されている.AlaRでは正荷電を持つArg136のグアニジド基が配置されているが,SerRで配位されたAsn84アミド窒素とフェノラート基O3の結合距離は,量子化学計算によるラセミ化反応シミュレーションにおいて2.8~3.0 Åの範囲にて終始固定されていた.セリンのラセミ化反応に伴う副反応として,PLPに収納できなかった電子が基質アミノ酸のβ位に流れ込みオーバーフローしたときに脱水反応が起こる.β位の水酸基は電子求引性が高く,小シンクの電子求引性と同等または上回ったときに脱水反応が起こるともいえる(図1E).
SerRにおけるPLPの電子状態は,ラセミ化反応を進めることはできるが,それでもα炭素に生じたカルボアニオンを安定化する電子シンクとしての容量が小さいことを示唆している.AlaRとSerRはどちらも二塩基機構によるラセミ化酵素であるが,PLPフェノラートO3の解離状態を制御する残基に着目するとArg(図1C)かAsn(図1E)という相違点があり,この違いが小シンクの電子求引性に影響している.Asn残基を配するSerRの方がイミド基の正電荷を弱めてしまうので,小シンクの電子吸引力は弱められて脱水反応をもたらすものと考えられる.この考察により,PLPの電子吸引性を決定するのはピリジン環Nに配位する残基(Asp, Arg, Ser/Thr)だけでなくO3に配する残基の影響も大きいといえる.
トレオニン合成酵素(ThrS)は,基質であるホモセリン4-リン酸との複合体を形成した後,まずリン酸が脱離してα-アミノクロトン酸とPLPとの複合体に変換した後に,その二重結合のβ炭素側に水分子を付加させてトレオニンを生成する(図1F).脱離したリン酸は活性中心にとどまり,β炭素に付加する水分子を固定する重要な役割を担う.SerRと同様にピリジン環Nには水酸基(Thr317),PLPの3位のフェノール基(−OH)にはアミド基(Asn87)と水分子も水素結合している.ThrSも,基質−PLPとそれに直接作用する周辺の5アミノ酸残基と4水分子を計算対象として高精度な量子化学計算が報告された10).
アミノクロトン酸とPLPの複合体からL-Thrとリン酸が生成物になる正反応,あるいはアミノクロトン酸が生成物として複合体から放出される副反応について,各素反応に関わる遷移状態の構造が計算に基づいて描かれ,自由エネルギーを縦軸とする反応ダイアグラムも描かれた.PLPの電子シンク機能を引き出すピリジンNに配するThr317水酸基,3位のフェノール基(−OH)に配する水との距離は反応シミュレーションの過程において固定しており変化しない.本報告では,遷移状態や中間体のHOMO/LUMO分子軌道の形状も報告されており,カルボアニオン中間体のHOMO軌道をみると基質のカルボキシル基からピリジン環Nまで広範な分子軌道が形成されていた.この分子軌道が示すことは,ThrRのPLPがピリジン環部分も活用してアニオン安定化に寄与していることである.水素結合による正電荷が大シンクによる電子求引性を示す一方で,Nに配位した水酸基のπ軌道電子がピリジン環に対して電子を供与するので,PLPはσ電子を基質から引き込みながらπ電子を基質側に押しだす難しい役割を果たしているといえる.PLP酵素の反応多様性を考察する上で分子軌道の概念を積極的に取り入れた新しい酵素化学の開拓が望まれる.
引用文献References
1) Kuwahara, M., Tamura, T., Kawamura, K., & Inagaki, K. (2011) Identification and conformer analysis of a novel redox-active motif, Pro-Ala-Ser-Cys-Cys-Ser, in Drosophila thioredoxin reductase by semiempirical molecular orbital calculation. Biosci. Biotechnol. Biochem., 75, 516–521.
2) Imada, K., Tamura, T., Takenaka, R., Kobayashi, I., Namba, K., & Inagaki, K. (2008) Structure and quantum chemical analysis of NAD(+)-dependent isocitrate dehydrogenase: Hydride transfer and co-factor specificity. Proteins-Structure Function and Bioinformatics, 70, 63–71.
3) 林秀行(2014)ピリドキサール酵素の反応機構,生化学,86, 214–231.
4) 吉村徹(2015)ビタミンB6酵素の構造,機能,進化に関する研究,ビタミン,89, 327–340.
5) Banik, S.D. & Chandra, A. (2014) A hybrid QM/MM simulation study of intramolecular proton transfer in the pyridoxal 5′-phosphate in the active site of transaminase: Influence of active site interaction on proton transfer. J. Phys. Chem. B, 118, 11077–11089.
6) Rubinstein, A. & Major, D.T. (2010) Understanding catalytic specificity in alanine racemase from quantum mechanical and molecular mechanical simulations of the arginine 219 mutant. Biochemistry, 49, 3957–3964.
7) Lin, Y.L., Gao, J., Rubinstein, A., & Major, D.T. (2011) Molecular dynamics simulations of the intramolecular proton transfer and carbanion stabilization in the pyridoxal 5′-phosphate dependent enzymes L-dopa decarboxylase and alanine racemase. Biochim. Biophys. Acta, 1814, 1438–1446.
8) Hayashi, H., Mizuguchi, H., & Kagamiyama, H. (1993) Rat liver aromatic L-amino acid decarboxylase:spectroscopic and kinetic analysis of the coenzyme and reaction intermediates. Biochemistry, 32, 812–818.
9) Xie, L., Liu, H., & Yang, W. (2004) Adapting the nudged elastic band method for determining minimum-energy paths of chemical reactions in enzymes. J. Chem. Phys., 120, 8039–8052.
10) Shoji, M., Hanaoka, K., Ujiie, Y., Tanaka, W., Kondo, D., Umeda, H., Kamoshida, Y., Kayanuma, M., Kamiya, K., Shiraishi, K., et al. (2014) A QM/MM Study of the L-threonine formation reaction of threonine synthase: Implications into the mechanism of the reaction specificity. J. Am. Chem. Soc., 136, 4525–4533.
著者紹介Author Profile
 田村 隆(たむら たかし)
田村 隆(たむら たかし)岡山大学大学院環境生命科学研究科農生命科学専攻生物機能化学講座教授.農学博士.
略歴1965年山口県に生る.88年岡山大学農学部卒業.93年京都大学大学院農学研究科修了,博士(農学).95~96年NIH滞在研究員.2011~14年さきがけ研究員.12年より現職.
研究テーマと抱負タンパク質の構造と機能に関する実験化学と計算科学研究.
ウェブサイトhttp://www.agr.okayama-u.ac.jp/ApplEnz/index.html
趣味外国語の習得,家事手伝い.