1)ミトコンドリアと品質管理機能
ミトコンドリアは真核生物に必須の細胞小器官(オルガネラ)である.電子伝達系を介して細胞に必要なATPの大部分を合成するという役割に加え,アミノ酸の生合成や脂質代謝,アポトーシス制御などの基盤となる.ミトコンドリアの起源は,およそ20億年前に原始真核生物が好気性細菌の一種であるプロテオバクテリアを取り込んだものと考えられている.この細胞内共生説は①ミトコンドリアが核とは異なる独自の環状DNAを有すること,②ミトコンドリアマトリクスに存在するリボソームは真核生物のものよりも細菌型のものに近いこと,③βバレル型膜タンパク質という原核生物の外膜に特徴的な膜タンパク質がミトコンドリア外膜にも見いだされること,などの証拠から広く浸透している.細胞内共生によって,酸素呼吸能を獲得した真核生物はその後,爆発的に進化を遂げたと考えられるが,一方でミトコンドリア自体はより多くのATPを合成し,宿主に供給しなければならなくなった.すなわち,ATP合成のための過剰な酸化的リン酸化反応の代償として,酸化ストレスの原因となる活性酸素種(ROS)の生成を余儀なくされることになった.ROSはタンパク質,脂質,DNAに損傷を引き起こす.そのため,ミトコンドリアに蓄積したROSにより,酸化的リン酸化反応系は機能不全に陥り,それによってさらにROSが生成されるという悪循環を招く.これに対応するためにミトコンドリアは,ROSの発生やそれに伴う局所的な機能不全に応答する品質管理機能を獲得していったと考えられる.たとえば,SOD2はミトコンドリアに局在するスーパーオキシドジスムターゼであり,活性酸素を無毒化する.また,ミトコンドリアマトリクスには種々のシャペロンやプロテアーゼが存在し,構造の不安定となったタンパク質のリフォールディングや分解を促進している.さらにミトコンドリアの障害が広範囲,つまりオルガネラ全体に広がった場合,損傷ミトコンドリアはオートファジーによって丸ごと排除される.
2)パーキンソン病とミトコンドリア障害
パーキンソン病(PD)は,中脳黒質のドーパミン作動性ニューロンが何らかの原因で変性・脱落することで引き起こされる神経変性疾患である1).日本国内では2番目に有病率の高い神経変性疾患であり,加齢に伴い発症するため,超高齢化社会を迎える日本にとってその発症メカニズムの理解と根本的な治療法の開発が急務となっている.PDは安静時振戦,筋強剛,運動緩慢,姿勢保持障害などの運動障害を主な症状とし,孤発性と特定の遺伝子変異に由来する遺伝性(家族性)のものに大別できる.PDの90%は孤発性であるが,その発症原因を特定することは困難を極める.PD患者において,ミトコンドリアの電子伝達系の活性が低下していることや,電子伝達系の阻害剤がPD様の症状を引き起こすという知見から,PDの発症原因にミトコンドリア機能不全が関与しているのではないかと考えられてきた.しかし,PDとミトコンドリア障害との明確な接点は永らく不明なままであった.この突破口となったのは2008年のRichard Youleらの報告である2).彼らは遺伝性PDの原因遺伝子産物の一つであるParkinが,膜電位の低下した損傷ミトコンドリアに特異的にリクルートされ,ミトコンドリアをオートファジー依存的な分解に導くことを実験的に証明した.彼らはミトコンドリアの膜電位を人為的に消失させる方法で,細胞に存在するほぼすべてのミトコンドリアがParkin依存的に分解されることを示した.実際のヒトの神経細胞でこのような極端な現象が起こるとは考えにくいが,簡便な方法で損傷ミトコンドリアの分解を追跡できるため,マイトファジーの研究は生化学・細胞生物学を中心として一気に花開き,その分子機構の解明は飛躍的に進んだ.細胞分裂がほとんど起きない神経細胞においては,ミトコンドリアの品質管理はきわめて重要と考えられる.現在では不良ミトコンドリアの選択的分解の機構が破綻し,神経細胞に不良ミトコンドリアが蓄積することで,パーキンソン病やアルツハイマー病などの神経変性疾患につながるというモデルが提唱されている.
本稿では,この10年あまりで加速度的に理解が進んだParkin依存性の損傷ミトコンドリア分解について紹介する.特にParkinとPINK1がどのように活性化し,損傷ミトコンドリアをオートファジーシステムで認識可能なカーゴへと変換させているか,その概要を発見の経緯を交えながら紹介したい.
3)マイトファジーの概略
マイトファジーは,ミトコンドリアがオートファジー依存的にリソソームで分解される現象と定義される.「マイトファジー」という用語そのものは2003年の文献からみられるようになった.一概にマイトファジーといっても,その誘導の条件や介在タンパク質はさまざまである.本稿では遺伝性潜性PD(正しくは遺伝性パーキンソン症候群とすべき)の原因遺伝子産物であるParkinとPINK1によって駆動されるマイトファジーに限定するので,その点,ご容赦いただきたい.
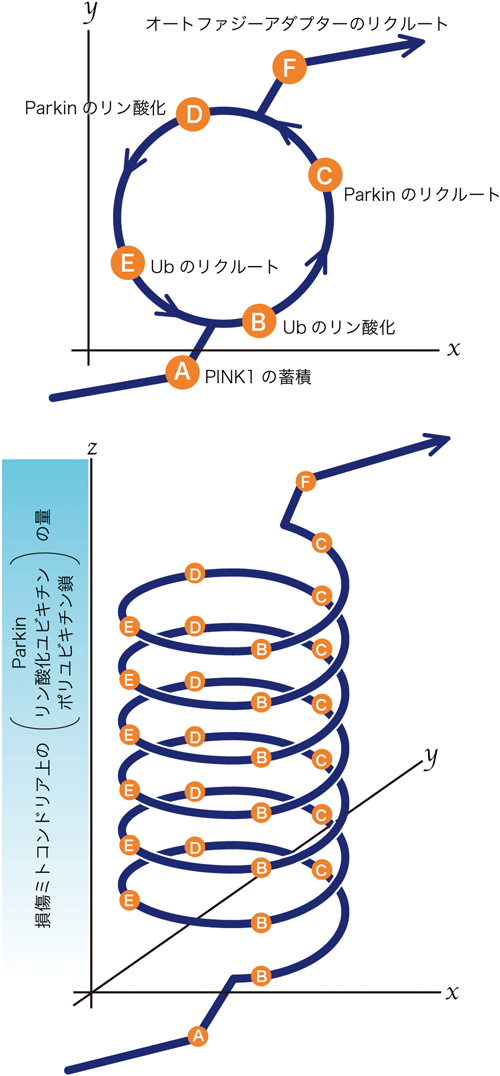
ParkinとPINK1はどちらも遺伝性潜性PDの原因遺伝子として,それぞれ1998年3)と2004年4)に同定された.Parkinはユビキチン分子を基質に付加するユビキチン連結酵素(E3リガーゼ)であり,PINK1はN末端にミトコンドリア移行シグナルを持つセリン・トレオニンキナーゼである.どちらも潜性遺伝であるため,ParkinとPINK1は「通常はパーキンソン病にならないようにプロテクティブに働く」と考えて妥当である.しかし,2008年まではParkinとPINK1の細胞内での機能はおろか,同定された基質に関しても研究者間でのコンセンサスは得られていなかった.前述のとおり,ParkinとPINK1の研究の大きな転機となったのは,Youleらによって報告された論文である2).彼らは,通常サイトゾルに局在しているParkinが,CCCPなどの脱共役剤でミトコンドリアの膜電位を消失させると,そこに選択的にリクルートされることを見いだした.さらにCCCPの長時間処理によって,オートファジー依存的にミトコンドリアがリソソームで分解されることを示した.現在では,ミトコンドリア膜電位の低下に伴いPINK1が外膜に蓄積し,ユビキチンのリン酸化を介して,サイトゾルのParkinが損傷ミトコンドリアにリクルートされると考えられている.そして損傷ミトコンドリア上にポリユビキチン鎖が形成され,オートファジーアダプターを橋渡し分子として,オートファジーマシナリーに認識されてリソソームで分解されるという,多段階を経た機構として理解されている(図1)(各ステップの分子機構の詳細は後述する).
2. ミトコンドリアの異常を検知する分子センサーPINK1
1)PINK1の分解と蓄積のメカニズム
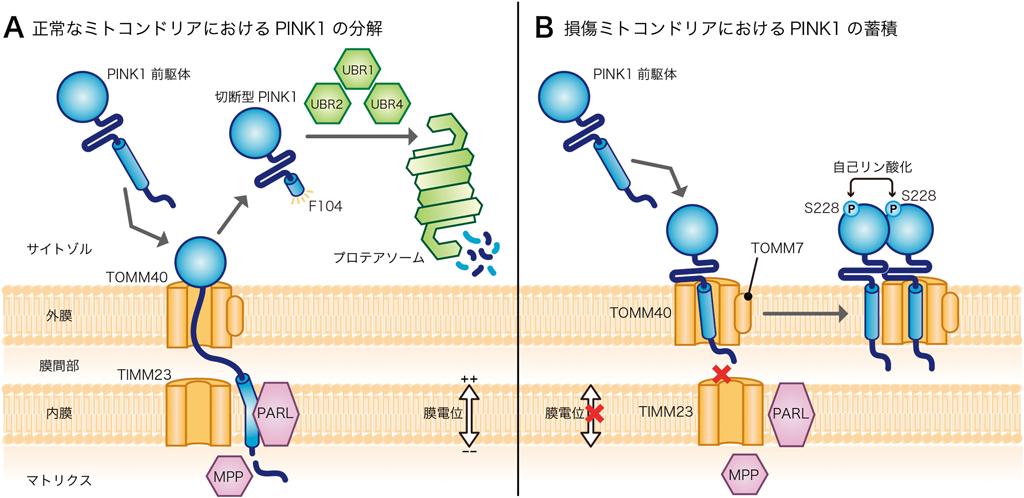
Parkinがマイトファジーの鍵因子であることが報告されてから,およそ2年後,PINK1がParkinの上流で機能することが,複数の研究グループから報告された5–9).ミトコンドリア異常を検知する分子センサーとも称されるPINK1は,(ヒトでは)581アミノ酸からなるセリン・トレオニンキナーゼであり,キナーゼドメインに加えて,N末端にミトコンドリア移行シグナル(プレ配列)と膜貫通ドメインを持つ.通常の培養条件,つまりミトコンドリアが正常な場合では,PINK1はサイトゾルのリボソームで合成後,直ちにミトコンドリア内に輸送される.ミトコンドリアの外膜・内膜にはTOMM40複合体,TIMM23複合体と呼ばれる膜透過チャネルが存在し,PINK1もその他の前駆体タンパク質と同様に,それらのチャネルを利用してミトコンドリアの中に入る.ただし,PINK1の場合はプレ配列部分がマトリクスに到達し,膜貫通ドメインが内膜に膜挿入されたトポロジーをとるとPARLと呼ばれるプロテアーゼによって切断される10).筆者らはPARLで切断されたPINK1(切断型PINK1)がミトコンドリアからサイトゾルに逆行輸送され,N末端則に従って,迅速にプロテアソームで分解されることを見いだした11).PARLによる切断とサイトゾルへの逆行輸送は,他の前駆体タンパク質ではみられないPINK1に特異的な現象である.したがって,ミトコンドリアが正常であれば,PINK1は半減期わずか15分以内で分解されてしまうユニークなキナーゼである(図2A).言い換えると,通常,PINK1はそのキナーゼとしての酵素活性を発揮せずに分解される.このようなPINK1の動態が不明のまま,それまでほとんどの研究者はPINK1を過剰発現した条件で,基質を探索していた.永らく研究者間でコンセンサスが得られなかったのはそのためであろう.
一方,脱分極剤処理によりミトコンドリア膜電位が消失すると,PINK1は外膜に蓄積する5, 9).PINK1のノックアウト細胞やキナーゼ活性のないPINK1変異体を発現させた細胞では,Parkinの損傷ミトコンドリアへのリクルートが完全に抑制されるため,PINK1はParkinの上流で機能するマイトファジーの必須因子である.では通常,迅速に分解されるPINK1がなぜ膜電位の消失で外膜に蓄積するのだろうか.これには,膜電位に依存した膜透過チャネルのゲーティングが鍵を握っている.ミトコンドリアの膜電位は内膜の膜透過装置TIMM23複合体のチャネルのゲーティングに必須であるため,膜電位の低下した不良ミトコンドリアではPINK1はTIMM23複合体を介して内膜に移行することができない.その代わりにPINK1はTOMM40複合体からラテラルに外膜に挿入される.筆者らは網羅的なsiRNAのスクリーニングから,この過程にはTOMM40複合体のサブユニットの一つであるTOMM7が必須であることを突き止めた12).さらにPINK1は外膜に組み込まれると二量体を形成し,自己リン酸化を介して活性化型のキナーゼとなる13, 14)(図2B).
2)PINK1はユビキチンをリン酸化する
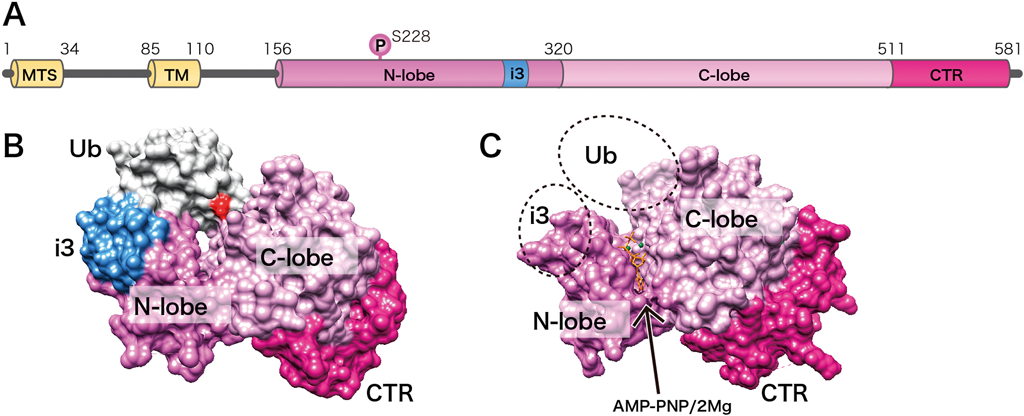
それではキナーゼとしてのPINK1の基質は何か.PINK1はParkinの上流で機能し,そのミトコンドリア移行に必須であるため,Parkinがその基質であると考えられた.実際,ParkinのN末端のUbl(Ubiquitin-like)ドメインがリン酸化されることがわかった15, 16).しかし,ParkinのUblドメインを欠失した変異体でも効率は悪くなるものの,完全にミトコンドリア移行を抑制することはできなかったため,PINK1の基質はParkin以外にも存在すると考えられた.そして,2013年に三つの研究グループが独立に,PINK1はユビキチンをリン酸化するキナーゼであることを報告した17–19).ParkinのUblドメインはユビキチン分子と非常に構造が似ており,リン酸化を受けるSer65も両者で保存されている.ユビキチンが,タンパク質のリシン残基に共有結合する翻訳後修飾因子であることは広く知られている.したがって,修飾する側であるユビキチン分子自身がリン酸化という翻訳後修飾を受けているという発見は,マイトファジーの研究分野のみならず,生化学・分子生物学全般に大きなインパクトを与えた.さらに最近,三つの研究グループからPINK1の立体構造が報告され20–22),PINK1がユビキチンの特異的なキナーゼであることが,構造生物学的アプローチからも確固たるものとなった.字数に制限があるため,個々の構造の詳細には言及できないが,以下はその要約となる.PINK1のキナーゼドメインはN-lobe, C-lobeから構成されており,その溝にATP結合部位を含む活性中心が存在する.ユビキチン分子あるいはATPの非加水分解アナログであるAMP-PNPとPINK1の共結晶構造から,ユビキチンのSer65近傍にATPが配置されている.他のキナーゼにはみられないPINK1に特徴的な挿入領域(インサート3)がユビキチン認識の要となっている.さらにPINK1の自己リン酸化部位がインサート3の正しい配置に必須であることも判明した(図3).
3. 損傷ミトコンドリアにユビキチンを付加するParkin
1)構造変化を伴うParkinの活性化
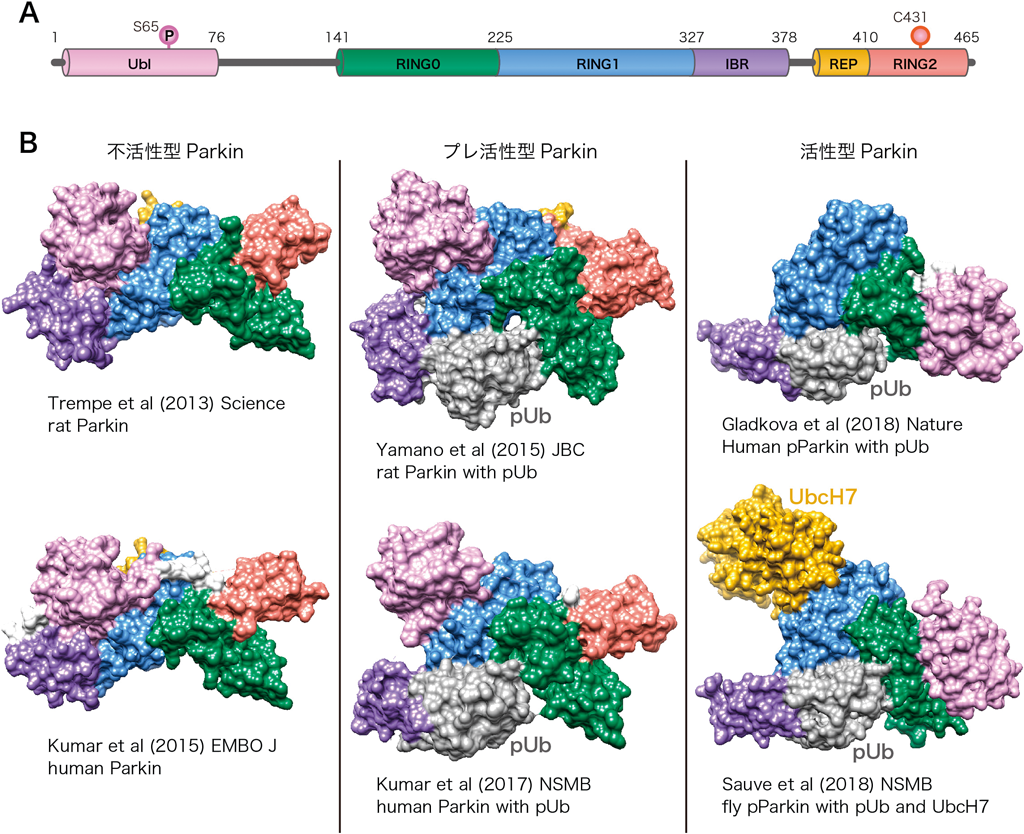
Parkinは複数のドメインから構成される(ヒトでは)465アミノ酸からなるE3リガーゼである(図4A).E3リガーゼはその構造からRING型,HECT型などに分類されるが,ParkinはRBR型リガーゼという特殊なE3に分類される.これまでRBR型はRING型の一種とされてきたが,RBR型リガーゼはユビキチン分子をRING2ドメインのシステイン残基にチオエステル結合で受け取ることができると報告された23).これはHECT型E3がユビキチンを授受する仕組みに類似しているため,RBR型はRING/HECTハイブリットとも呼ばれる.興味深いことにRBR型E3はヒトゲノムに13種類しかコードされていない.RING型E3が500種類以上あることを考えると,RBR型がいかに特殊であるかをわかっていただけると思う.Parkinの全長の立体構造は,2013年にTrempeらによって報告された24)(図4B).ParkinはN末端からUbl, RING0, RING1, IBR, REP, RING2ドメインによって構成されるが,この構造からParkinが普段は不活性型のE3であることが再認識された.RBR型E3リガーゼは,基質のユビキチン付加のためにE2酵素からユビキチンを受け取り,自身のシステイン残基にロードすることが必須である.しかし,Parkin単独の構造はRING1ドメイン上のE2相互作用面が,REP(repressor element of Parkin)と呼ばれる短いαヘリックスによってマスクされていること,また,ユビキチン分子を受け取るRING2ドメイン上のシステイン残基(Cys431)が構造内部に埋もれていることが判明した.前述のとおり,Parkinはリン酸化ユビキチンとUblのリン酸化によって活性化型E3に変換される.そこで筆者らは,まず,リン酸化ユビキチンがParkinと高い親和性で相互作用することを確認した.さらに光架橋性側鎖を持つ非天然アミノ酸を部位特異的にリン酸化ユビキチンとParkinに導入し,相互作用マッピングを行うことで,ParkinのRING0とRING1ドメインのまたがる領域にリン酸化ユビキチンがはまり込むことを明らかにした25)(図4B).ほぼ同時期にParkinとリン酸化ユビキチンの共結晶構造も解かれ26),同様の結論に至る報告が相次いだ27–29).リン酸化ユビキチンがParkinに結合すると,構造変化が誘起され,それまで強固に相互作用していたUblがコアから外れる.それをPINK1が認識してUblのSer65をリン酸化する.最近,リン酸化Parkinの結晶構造も明らかとなり,Parkinの活性化メカニズムにさらなる進展がみられた30–32)(図4B).驚くべきことにUblがリン酸化を受けると,(それまでRING2と相互作用していた)RING0に結合することがわかった.つまり,リン酸化UblがRING0に結合することで,RING2をRING0による抑圧から外している.これはParkinがE2からユビキチンを受け取ることのできる活性型E3になったことを意味している.
2)増幅される損傷ミトコンドリア上のユビキチン鎖
上述のように,何らかのストレスで細胞内に損傷ミトコンドリアが発生すると,そこにPINK1が蓄積し,ユビキチンをリン酸化する.そして,リン酸化ユビキチンとの親和性で,サイトゾルのParkinが損傷ミトコンドリアにリクルートされることはおわかりいただけたと思う.ではPINK1やリン酸化ユビキチン以外に,Parkinのミトコンドリア移行に必要な因子はあるのだろうか.実はParkin自身のE3活性もミトコンドリア移行に必要である33–35).ParkinのE3活性のない変異体では,損傷ミトコンドリアへのリクルートはほぼまったく観察されない.またE1阻害剤の添加により,ユビキチンシステムの機能を根本から破綻させた場合でもParkinのリクルートが阻害される.さらにリン酸化ユビキチンの存在量に関しても興味深い報告がある.Parkinを発現していないHeLa細胞では脱分極剤の処理を行っても,リン酸化ユビキチンはほとんど合成されない.一方,Parkinを過剰発現させると,リン酸化ユビキチンの存在量はユビキチン全体の3%(脱分極剤の処理時間等の違いで,報告によってその値は多少異なる)まで上昇する.つまり,リン酸化ユビキチンの合成量は,その下流で機能するParkinに依存していることになる.このような実験結果をもとに,現在ではParkinのミトコンドリア移行はポジティブフィードバックユビキチン化サイクルによって駆動される35, 36)と考えられている(図5).PINK1の蓄積によってミトコンドリア外膜で生成されるリン酸化ユビキチンは非常にわずかな量であり,それを受容体としてリクルートするParkinもごくわずかである.しかし,このParkinはUblのリン酸化を介して活性化し,ミトコンドリアタンパク質をユビキチン化する.このユビキチン化は,サイトゾルのユビキチン分子がミトコンドリアにリクルートされることを意味するので,新たにミトコンドリア外膜にリクルートされたユビキチンはPINK1のリン酸化のターゲットとなる.その反応で生成されたリン酸化ユビキチンは,さらにサイトゾルのParkinをリクルートするシードとなる.つまり,このサイクルが回ることで,PINK1の蓄積量はわずかでも,損傷ミトコンドリアには膨大な分子数のParkinがリクルートし,損傷ミトコンドリアは高度にユビキチン化されることになる.原理的にはサイトゾルのParkinまたはユビキチン分子がなくなるまで,あるいは損傷ミトコンドリアがオートファゴソームで隔離されるまでこの反応は続くと考えられる.また,この反応は損傷ミトコンドリアに限定されて起こるため,同じ細胞の正常なミトコンドリアはその影響を受けない2, 5).
3)Parkinの基質選択性
上述のようなユビキチン化反応のサイクルによって,損傷ミトコンドリアの外膜タンパク質にはユビキチン鎖が付加される.質量分析を利用した網羅的な解析から,Parkinはさまざまなミトコンドリア外膜タンパク質にポリユビキチン鎖を付加できることがわかっている37).つまり,損傷ミトコンドリア上で活性化したParkinは,近傍のタンパク質であれば無差別にユビキチン鎖を付加し,その基質選択性はきわめて低いと考えられる.マイトファジーの場合,後述するようにユビキチン鎖がオートファジー認識の標識となるので,効率的なミトコンドリア分解という観点から考えると,損傷ミトコンドリアという選択性さえあれば,そこに含まれている基質の選択性は低い方が都合がよいのかもしれない.しかし一方で,特定の基質にユビキチンを付加することにより,マイトファジーを正に制御することも報告されている.たとえば,ミトコンドリアの融合を促すGTPaseであるMFN1やMFN2がそれに該当する38).マイトファジーの刺激によって,損傷ミトコンドリアのMFN1やMFN2が迅速にユビキチン化を受ける(あるいはそれによりプロテアソームで分解される)と,損傷ミトコンドリアは正常なミトコンドリアと融合できなくなる.つまり,MFN1やMFN2のユビキチン化は,不良ミトコンドリアの隔離に寄与しているのかもしれない.また,Miro1と呼ばれるGTPaseは,微小管上のキネシンモータータンパク質と結合することで,細胞内のミトコンドリアの輸送を制御している.神経細胞においても,ミトコンドリアは常に細胞体–軸索末端間を行き来しているが,リソソームは細胞体に多く存在する.Parkin依存的なMiro1の迅速な分解は,損傷ミトコンドリアの軸索末端への移動を抑制し,リソソームの多く存在する細胞体への輸送を促進していると考えられる39).
4)他因子によるユビキチン付加の制御
Parkin分子だけでマイトファジー誘導に必要十分なユビキチン鎖が形成されるかは不明である.Parkinによって損傷ミトコンドリア上のユビキチン鎖の大部分が合成されることは間違いないと考えられるが,ミトコンドリア外膜に局在する,あるいはサイトゾルに局在する他のE3リガーゼの寄与も否定できない.特に,上述のポジティブフィードバックユビキチン化サイクルが正しいとすると,PINK1によってリン酸化される最初のユビキチン分子はどのようにミトコンドリア外膜に局在するのであろうか.最上流のリン酸化ユビキチンはParkinが活性化される前にミトコンドリアに局在すべきであり,そしてそれはParkin非依存的に起こるはずである.ミトコンドリア外膜タンパク質はマイトファジー非誘導時でもユビキチン・プロテアソーム系で分解を受け,合成と分解のバランスを保っているので,そのような「たまたま」外膜タンパク質に結合していたユビキチンをPINK1が利用するかもしれない.あるいは,Parkinの上流で損傷ミトコンドリアにユビキチンを付加する未知のシステムがあるのかもしれない.
さらに,ユビキチン鎖は脱ユビキチン化酵素(DUB)によって基質から除去されるため,DUBはマイトファジー調節に寄与している40, 41).特にミトコンドリア外膜にアンカーされているUSP30は,Parkinによって外膜に形成された特定のユビキチン鎖を選択的に除去することで,マイトファジーを負に制御している42–44).
1)オートファジーの膜動態について
マイトファジーを誘導すると,損傷ミトコンドリア近傍に隔離膜と呼ばれる膜構造が出現する.隔離膜は経時的に損傷ミトコンドリアに沿ってカップ状に伸長し,最終的に縁が閉じたオートファゴソームと呼ばれる二重の膜構造体となる.その後,オートファゴソームはリソソームと融合し,内包物(マイトファジーの場合は損傷ミトコンドリア)が分解される.オートファジー必須タンパク質(コアATGタンパク質)は酵母からヒトまで進化的に保存されているものが多く,それらはいくつかの機能ユニットに大別できる.ただし,オートファゴソームが完成すると,ほとんどすべてのコアATGタンパク質はそこから遊離する.唯一,完成したオートファゴソームにも局在できるのが(出芽酵母では)Atg8と呼ばれるタンパク質である45).Atg8は,ユビキチンシステムに類似の機構で脂質分子であるホスファチジルエタノールアミンと共有結合するため,オートファゴソーム膜に直接アンカーできる.哺乳動物には酵母Atg8のホモログが6種類あり,LC3A, LC3B, LC3C, GABARAP, GABARAPL1, GABARAPL2と呼ばれている.隔離膜あるいはオートファゴソーム上のAtg8の機能は種々提唱されているが,選択的オートファジーにおいては,分解基質の取り込みにとりわけ重要であろう.Atg8には疎水性のアミノ酸残基で構成されるくぼみがあり,このくぼみにはまり込むことができるペプチド配列はAtg8と相互作用できるため,そのような配列を持つタンパク質は,オートファゴソームに取り込まれる分解基質になりうる46).この配列は弱いコンセンサス配列(W/Y/FxxL/I/V)を持ち,LIR(LC3 interacting region)あるいはAIM(Atg8-family interacting motif)と呼ばれる47).
2)ユビキチンとオートファジーを橋渡しするアダプター
損傷ミトコンドリアにユビキチン鎖が付加されたことをオートファジーシステムに伝えるのは,オートファジーアダプターと称される分子群である48).オートファゴソームは損傷ミトコンドリアを認識する選択性を持たないので,オートファジーアダプターがその役割を担っている.オートファジーアダプターはその分子内にユビキチン結合ドメインとLIRの両方を有するので,それを橋渡しとして,ユビキチン化された損傷ミトコンドリアはオートファゴソームに囲まれる.マイトファジーの場合,これまでにOPTN, NDP52, p62, NBR1, TAX1BP1の少なくとも5種類のオートファジーアダプターが同定されている.そして,これら五つのアダプターはすべて,ユビキチン鎖依存的に損傷ミトコンドリアにリクルートされることがわかっている.五つすべてのオートファジーアダプターをノックアウトした培養細胞の実験から,どうやらマイトファジーにはOPTNとNDP52の二つが特に重要であるらしい49, 50).さらにTBK1というキナーゼがオートファジーアダプターのユビキチン結合ドメインやLIRやその近傍をリン酸化することで,それらのアフィニティーを調節している50, 51).
3)損傷ミトコンドリア上のユビキチン鎖を認識するエンドソーム因子
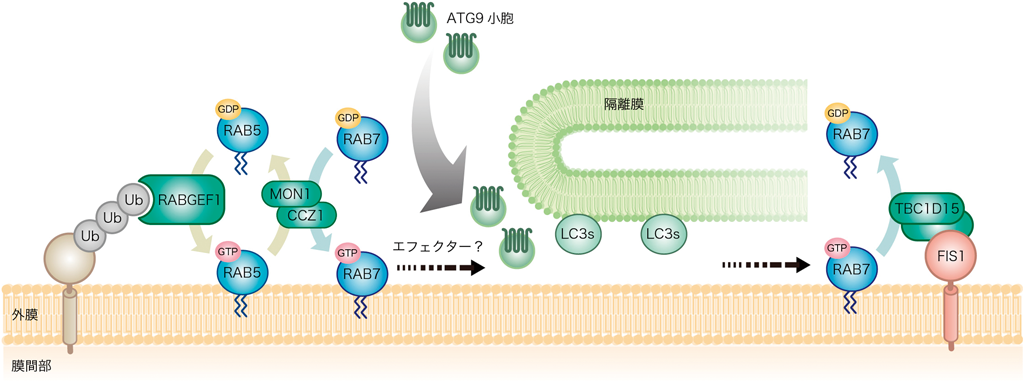
ミトコンドリアは分裂・融合を絶えず繰り返す,非常にダイナミックな動態を示すオルガネラである.上述したように,ミトコンドリア融合を担うMFN1およびMFN2は,マイトファジーの誘導でいち早く分解される.さらに,ミトコンドリア分裂を促進するダイナミン様タンパク質DRP1も効率的なマイトファジーに必要であると報告されている.また,FIS1は酵母からヒトまで進化的に保存されているミトコンドリア外膜のタンパク質であり,出芽酵母のFis1はDnm1(DRP1の酵母ホモログ)をミトコンドリアへとリクルートする受容体である.そのため,ヒトFIS1も長い間,形態制御因子であると考えられてきた.しかし,ヒト培養細胞でFIS1遺伝子をノックアウトしてもミトコンドリア形態に明確な異常がみられないこと52),DRP1の受容体として,哺乳類でMff, MiD49, MiD51が同定されたこと52, 53),ヒトFIS1の相互作用パートナーとしてTBC1D15が同定されたこと54)などにより,FIS1の形態制御機能に疑問が呈された.そんな折,偶然にも筆者らは,FIS1を欠損した線虫ではLGG-1(Atg8の線虫ホモログ)が異常に蓄積することを見いだした55).このLGG-1の蓄積はミトコンドリアストレスによって増大し,PINK1の発現抑制で減少したため,FIS1のマイトファジーへの関与が疑われた.筆者らはFIS1とその相互作用因子であるTBC1D15(後期エンドソームやリソソームに局在するsmall GTPase RAB7Aの活性化因子)の機能解析から,哺乳類培養細胞でもマイトファジー依存的かつFIS1あるいはTBC1D15依存的に,オートファゴソームマーカーであるLC3Bが異常に蓄積することを見いだした56).さらに解析を続けた結果,最終的に,エンドサイトーシスの一連のタンパク質が効率的なマイトファジーに重要であるという結論にたどりついた57).以下にその要約を示したい.マイトファジーの刺激により,損傷ミトコンドリア上にユビキチン鎖が形成されると,エンドサイトーシスの上流で機能するユビキチン結合タンパク質であるRABGEF1が損傷ミトコンドリアにリクルートされる.これによって,下流のRAB5, MON1/CCZ1複合体,RAB7が連鎖的にリクルートされ,オートファジーシステムを活性化する.特にRAB7のノックダウンによりATG9と呼ばれるオートファジー因子のミトコンドリア近傍へのリクルートが阻害される.ATG9はオートファジー必須因子の中で唯一,複数回の膜貫通ドメインを有するタンパク質であり,ゴルジ体由来の膜小胞に組み込まれている.詳細はいまだ不明な部分も多いが,マイトファジー誘導時のATG9小胞の動態は,RAB7を介するメンブレントラフィックによって制御されるものと予想される.TBC1D15はRAB7の特異的なGAP(GTPase activating protein)として機能するため,その発現(あるいはTBC1D15のミトコンドリア局在に必須のFIS1の発現)を抑制すると,RAB7はGTP型からGDP型に変換できずに常にスイッチオンの状態になる.線虫や哺乳類培養細胞でみられた無秩序なオートファゴソームの蓄積(まるで隔離膜が正しくミトコンドリアを囲い込むことができずに暴走してみえる現象)は,RAB分子スイッチの制御不能によって引き起こされたと解釈できる(図6).
Parkin依存性のマイトファジーは,膜電位の低下したミトコンドリアのみを選択的に分解する洗練された品質管理機構であり,ユビキチンとオートファジーの細胞内2大分解系が協調的に働くシステムである.また,ParkinとPINK1がPDの原因遺伝子であるため,神経科学や臨床医学からも注目されている.したがってマイトファジーはこれら異分野研究者が参入する非常に競争の激しい研究領域となっている.本稿ではこの10年あまりで明らかとなったParkin依存性マイトファジーの生化学・細胞生物学・構造生物学の知見を中心に紹介させていただいた.スペースの都合上,生理学的意義解明に向けた動物個体を用いた研究など,多くの重要なトピックについて紹介しきれなかったことをお詫びしたい.
一般的にミトコンドリアタンパク質は,シグナル配列の付加によってミトコンドリアに局在することが運命づけられている.その配列がどういう特異性を持っているか,ミトコンドリアの受容体や膜透過チャネルはどのような機能で前駆体タンパク質の膜透過や膜挿入を実現させているか,これが筆者の修士から博士,最初のポスドク2年間の研究テーマであった.8年間没頭したミトコンドリアタンパク質の輸送の研究から卒業し,次の研究テーマを探していた,そんな折にYouleらによって報告されたParkin依存性マイトファジーの論文は衝撃的だった.多くの研究者は,PD原因遺伝子産物であるParkinの機能がミトコンドリア分解にあることに魅了されたと思うが,筆者はParkinのミトコンドリア移行にとりわけ興味が湧いた.ミトコンドリアタンパク質の輸送には膜電位が必要(正しくは内膜を通過する,あるいは内膜に組み込まれる場合に必須)であるのに,Parkinはなぜか膜電位の低下したミトコンドリアに局在する.しかも普段はサイトゾルに局在しているのに,ミトコンドリアの品質でその局在が変化する.これまでのミトコンドリアタンパク質の輸送の概念からは到底説明できないものであった.そこで,この分子メカニズムを解明したいとの思いで,Youleの研究室に留学することを決意した.結局,Parkinのミトコンドリア移行およびマイトファジーを駆動しているのはPINK1前駆体の膜電位依存的な局在変化であったから,筆者にとっては運命的なものを感じずにはいられない.とにもかくにも,これがきっかけとなり,ユビキチンやオートファジーによって制御されるミトコンドリア品質管理に研究の焦点をシフトしていった.今後は,マイトファジーの破綻が生物個体にどのような影響をもたらすかについて,勢力的に研究が展開していくと予想されるが,マイトファジーの本質の理解にはまだまだ生化学・分子生物学的アプローチに頼るところが大きいはずだ.