アルポート症候群は,腎臓の糸球体の基底膜を構成するType IV collagen遺伝子(COL4A3, COL4A4, COL4A5)のいずれかの変異を原因とする遺伝性疾患である.多くが小児期から発症し,10~20代で末期腎不全に至る重篤な疾患である1).現在,アルポート症候群の治療は,他の慢性腎臓病と同様にレニン・アンジオテンシン系(RAS)阻害剤による対症療法が行われる2).糸球体腎炎では,糸球体内圧の低下を企図した降圧剤による治療が一般的であり,アルポート症候群の治療に関しても例外ではない.一方,アルポート症候群患者の予後は,RAS阻害剤による早期治療介入により改善が認められるものの,最終的に例外なく末期腎不全へ進行し,人工透析もしくは腎移植を余儀なくされることとなる.したがって,現在の対症療法に加えて,病気の発症機序に基づく直接的な治療法の開発が強く求められている.
そのような背景の中,筆者らはアルポート症候群の原因タンパク質COL4A3/A4/A5の喪失した機能の正常化による本疾患の根本治療法の可能性を検討してきた.そして,最近,アルポート症候群の原因タンパク質の機能をハイスループットに評価できる系の構築に成功し,原因を標的とした治療薬の開発に一歩近づいた.本稿では,筆者らが開発したアルポート症候群の原因タンパク質COL4A3/A4/A5の機能評価系3)を概説し,本疾患に対する治療薬開発の可能性について議論したい.
2. アルポート症候群とType IV collagen
腎臓の糸球体基底膜は主にType IV collagen α3α4α5, Laminin-521(α5/β2/γ1),Nidogen, Agrinによって構成されている4).アルポート症候群は,Type IV collagen α3α4α5をコードするCOL4A3/A4/A5遺伝子の変異により,Type IV collagen α3α4α5による基底膜の形成ができなくなることで,糸球体基底膜の恒常性が失われ,最終的に糸球体の尿濾過機構が破綻することが発症原因である1).
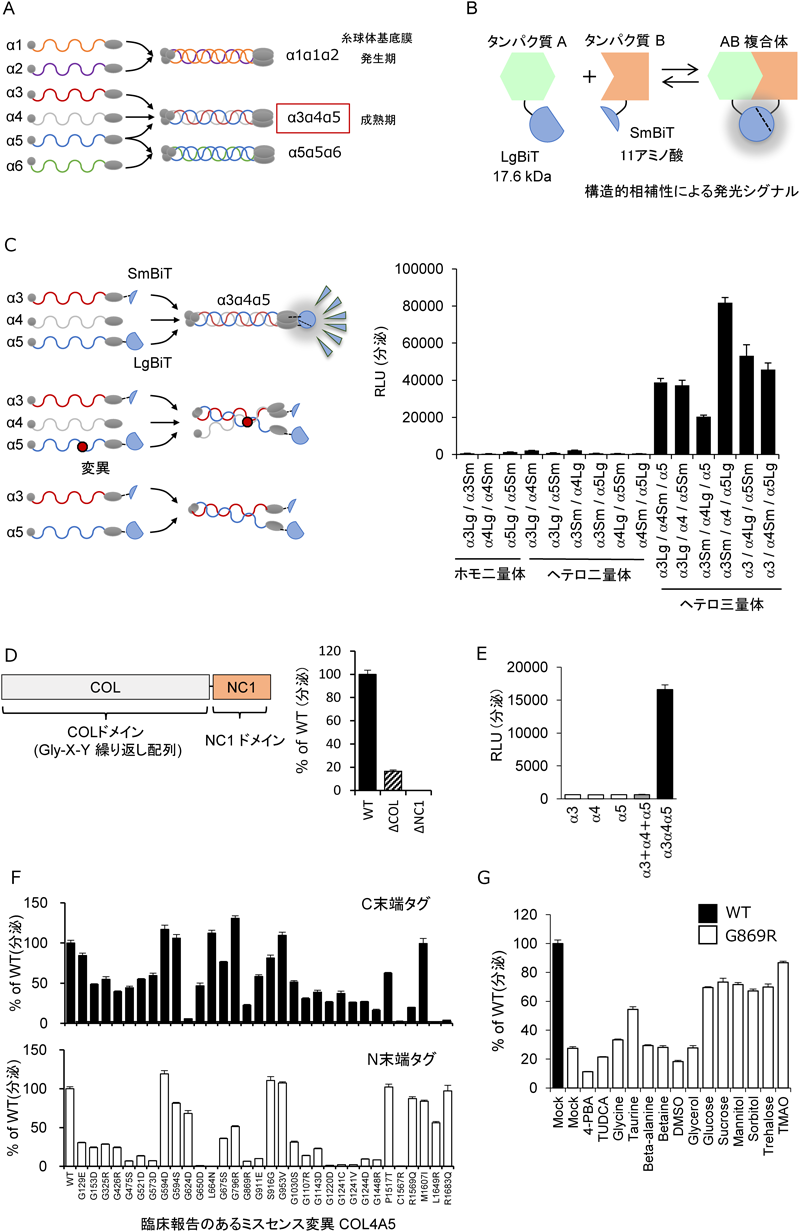
Type IV collagenは,6種類のα鎖(α1~α6)から決まった組合わせの三量体(α1α1α2, α3α4α5, α5α5α6)として細胞内で形成され,分泌される(図1A).分泌された三量体は細胞外でそれぞれhead-to-head, tail-to-tailで結合し,六量体を形成することで基底膜のネットワークを形成する.腎糸球体基底膜では発生期にType IV collagen α1α1α2による基底膜が発現し,発生後期にはα3α4α5へと構成因子が変化することで糸球体基底膜の維持が行われている.一方,アルポート症候群では,COL4A3/A4/A5遺伝子変異によりType IV collagen α3α4α5を含む糸球体基底膜の形成不全が引き起こされる.このとき,本来は主に発生期に発現し成熟糸球体では消失するはずのα1α1α2が発現し続けることで糸球体基底膜が維持されるが5),代償性α1α1α2の発現だけでは糸球体基底膜の恒常性を長期的に保つことができない.この要因として,α1α1α2が形成するネットワークはα3α4α5のものと比べて,分解シグナルに対して脆弱であることが想定されている.事実,ジスルフィド結合(S–S)の数はα1α1α2と比べてα3α4α5が多い6).これらのことから,アルポート症候群の発症原因に基づく治療法の開発において,変異α3α4α5の機能の回復による糸球体基底膜の恒常性維持が新たな治療標的となりうることが想定される.
3. Type IV collagen α3α4α5の機能の回復
筆者らはアルポート症候群のCOL4A5遺伝子の変異の中でもミスセンス変異に着目して,変異によって失われるType IV collagen α3α4α5の三量体形成能を是正することができればアルポート症候群の原因に基づく画期的な治療法の開発になると考えた.
1)変異COL4A5の細胞内安定化による治療の可能性
変異を有するCOL4A5は,α3, α4とのヘテロ三量体形成能を低下させ,機能的なType IV collagen α3α4α5の産生を減少させると考えられている.しかし,①変異COL4A5の細胞内安定性が低下した結果,α3α4α5三量体が作られなくなるのか,②変異COL4A5は細胞内で安定性を保っているが,三量体が形成できないのか,は明らかでなかった.もし,前者の細胞内安定性が問題であれば細胞内分解を抑制することによってα3α4α5三量体を増加させることができると考えられる.そこで,各種ミスセンス変異COL4A5(G869R, G1107R, P1517T, C1567R, L1649R)を293T細胞に過剰発現し,タンパク質安定性を評価した.その結果,野生型および変異COL4A5の細胞内安定性に有意な変化はなく,ミスセンス変異COL4A5は細胞内安定性を野生型と同等に保つことを明らかにした.したがって,アルポート症候群におけるα3α4α5ヘテロ三量体形成の回復には,変異COL4A5の細胞内安定化ではなく異なる標的が必要であることが明らかとなった.
2)変異COL4A5の小胞体シャペロンによる細胞内局在制御による治療の可能性
前述のように,ミスセンス変異を有するCOL4A5は野生型と同等のタンパク質安定性を有することが明らかになった.そこで,野生型と変異COL4A5の小胞体シャペロンによる細胞内局在制御機構に差異があるか否かに関して検討を行った.野生型と変異COL4A5(G869R, C1567R)を発現する細胞に代表的な小胞体シャペロン(BiP, GRP94, PDI, CRT, HSP47)を過剰発現およびノックダウンし,細胞内と細胞培養上清中のタンパク質発現量の変化を検討した.その結果,各種小胞体シャペロンの発現によってCOL4A5の細胞内局在が制御されるものの,野生型と変異COL4A5における顕著な差異は認められなかった.したがって,変異COL4A5の機能喪失を是正するためには,細胞内安定性や細胞内局在制御ではなく,α3α4α5ヘテロ三量体の形成そのものを標的化することが必要であることが示唆された.
3)COL4A5のα3α4α5ヘテロ三量体形成評価系の開発
Type IV collagen α3α4α5の三量体形成を標的とした治療法の確立には,α3α4α5ヘテロ三量体を評価する系の構築が必要である.これまでに,免疫沈降法によってα3α4α5ヘテロ三量体の評価が可能であることが報告されているが7–9),実際の創薬への応用性を考えると,三量体形成を亢進しうる化合物の探索に適したハイスループット性を有するα3α4α5ヘテロ三量体評価系の開発が求められる.将来的な化合物スクリーニングへの適応を見据えて,本研究室では新たな定量的かつ高感度にα3α4α5ヘテロ三量体を検出できる評価系の開発に関して種々の検討を行った.
ハイスループットなα3α4α5ヘテロ三量体評価系の確立のため,本研究室では,split NanoLuciferase(split NanoLuc)によるタンパク質相互作用評価系を用いた10)(図1B).その理由として,split NanoLucは,NanoLucの断片を付加したタンパク質どうしの相互作用を発光により検出する系であり,他のタンパク質相互作用を検出する方法と比較して高感度であること,NanoLucは他の発光・蛍光タンパク質と比較してサイズが小さいことから(19 kDa),巨大分子であるType IV collagen(単量体:約180 kDa,三量体:約540 kDa)の本来の三量体形成に与える影響が少ないことが予想されたためである.
そこで,まず,NanoLuc断片融合タンパク質の最適化を行った.さまざまな組合わせでsplit NanoLuc断片(LgBiT, SmBiT)をそれぞれα5, α3のC末端に付加し,標識されたα鎖をホモ二量体(α3-LgBiT/α3-SmBiT, α4-LgBiT/α4-SmBiT, α5-LgBiT/α5-SmBiT),ヘテロ二量体(α3-SmBiT/α4-LgBiT, α3-SmBiT/α5-LgBiT, α4-SmBiT/α5-LgBiT),ヘテロ三量体(α3-SmBiT/α4/α5-LgBiT)を作りうる組合わせで293T細胞に過剰発現し,培養上清中の発光を測定した.その結果,α3α4α5ヘテロ三量体を形成するすべての条件において発光を確認し,一方,α3α4α5ヘテロ三量体を形成しえないようなホモ二量体,ヘテロ二量体の条件で過剰発現した細胞の培養上清中の発光は認められなかった(図1C).そこで,最も効率のよいsplit NanoLuc断片の組合わせを用いて,N末端付加による三量体の検出を検討したところ,C末端付加と同様にα3α4α5ヘテロ三量体を形成する条件でのみ培養上清中の発光が顕著に認められた.これらの検討から,Type IV collagen α3/α4/α5のCおよびN末端に融合したNanoLuc断片はα3α4α5ヘテロ三量体が会合したときのみ近接し,基質の存在下で発光することが示唆された.
次に,認められた発光がα3α4α5ヘテロ三量体の形成量依存的であるか検討を行った.α3-SmBiT/α4/α5-LgBiT共発現において発光検出される発光は,三量体の構成因子の一つであるα4発現量依存的に増加すること,非標識α3/α5の発現により競合的に発光が減弱することから,本評価系は,α3α4α5ヘテロ三量体特異的に検出できることが示唆された.
Type IV collagenには,二つの構造的に重要なドメインが存在する.一つ目はNC1ドメインと呼ばれ,collagenのC末端にある非らせん構造をとり,三量体形成の初期会合に重要である.二つ目は,COLドメインと呼ばれ,アミノ酸のGly-X-Yを基本配列とした繰り返し配列で,collagenの特徴的な三重せん構造の形成に重要なドメインである11).したがって,これらを欠失したCOL4A5は三量体を形成しないことが想定される.そこで,ドメインの欠失変異COL4A5(ΔCOL, ΔNC1)を用いたところ,顕著な三量体形成抑制が認められた(図1D).さらに三量体形成に伴い認められる発光がType IV collagenの生理的な制御機構を反映しているかについて種々の検討を行った.多くのcollagenの生合成に重要な補酵素アスコルビン酸の処理12, 13)は本評価系における三量体の分泌を増加させた.また,各α鎖単独発現細胞(α3/α4/α5)の共培養(α3+α4+α5)では発光は認められず,すべてのα鎖を共発現した場合(α3α4α5)においてのみ発光が認められたことから,Type IV collagen α3α4α5は細胞内で複合体を形成し,分泌される特性を反映していることが確認された(図1E).
次に,臨床報告があり,頻度の高いCOL4A5ミスセンス変異の約30種類に関して14),細胞内で三量体が形成し,細胞外へ分泌された三量体の量を指標に,各種変異COL4A5の三量体形成能を評価した.その結果,全変異体のうち,約4割の変異体がC末端およびN末端タグ評価系でともに野生型の50%以下に発光の減少を認めた.C末端もしくはN末端のいずれかで発光の減少が認められた変異体は評価した変異体のうち約8割であった.したがって,本評価系は,CおよびN末端タグの二つの評価系を用いることでα5変異による機能喪失の多くを反映できることが示唆された(図1F).一方,一部の変異体はどちらの評価系においても発光の減少を認めなかった.これら変異については三量体形成能が保持されている可能性があり,糸球体基底膜上のType IV collagen α 3α 4α 5発現との相関をさらに検討していく必要がある.さらに細胞内と細胞外の複合体形成を比較したところ,半数以上の変異COL4A5が細胞内ではType IV collagen α3α4α5三量体を形成するものの,分泌不全であることが示唆された.
4)化合物による変異COL4A5の三量体形成促進による治療の可能性
最後に,Type IV collagen α3α4α5三量体評価系を用いて変異COL4A5の喪失した三量体形成能を化合物により是正することが可能であるか否か検討した.まず,頻度の高いG869R変異を代表例とし,ケミカルシャペロン様作用がすでに報告されている化合物14種類15)を処理した.その結果,浸透圧調節系のケミカルシャペロンによりG869R変異COL4A5の三量体形成に伴う分泌の増加が認められた(図1G).高い効果が認められたMannitolおよびTMAOに関して,他の変異についても同様に効果を検討したところ,Gly-X-YであるCOLドメイン上のGly置換変異体G1107R, G1143D, G1244Dに対しても三量体形成の増加を示した.以上のことから,低分子化合物により変異COL4A5の三量体形成および分泌を是正できる可能性を明らかにした.