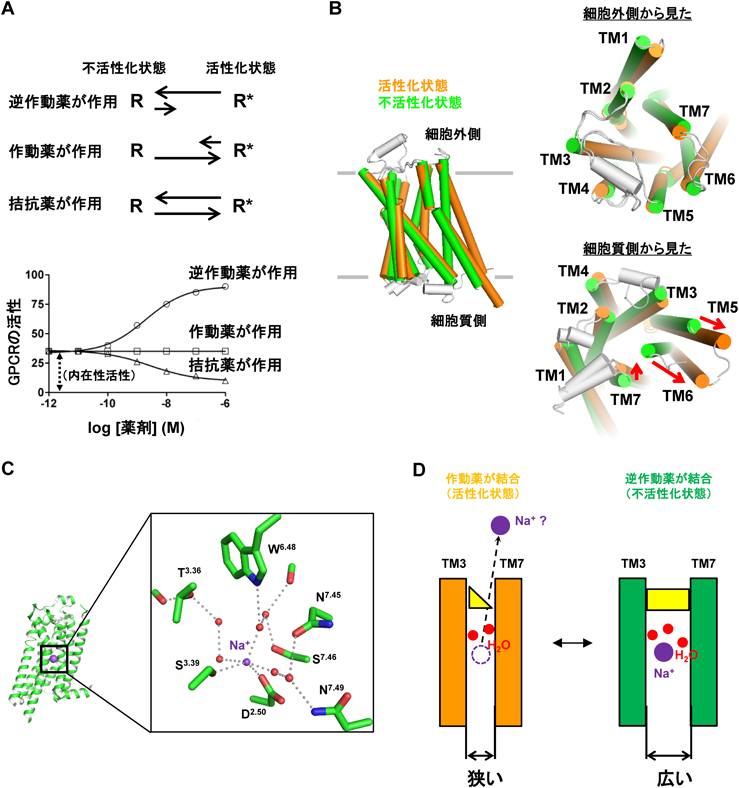
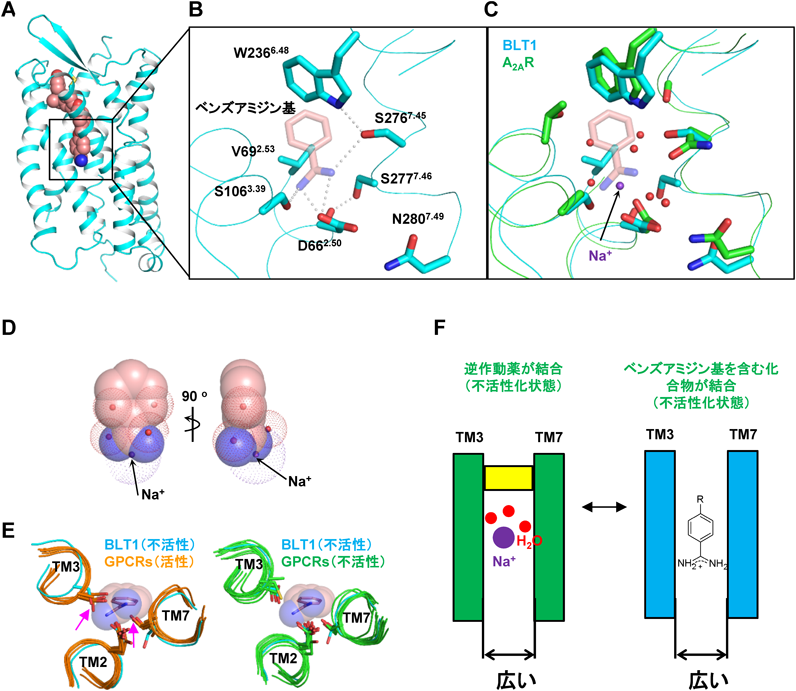
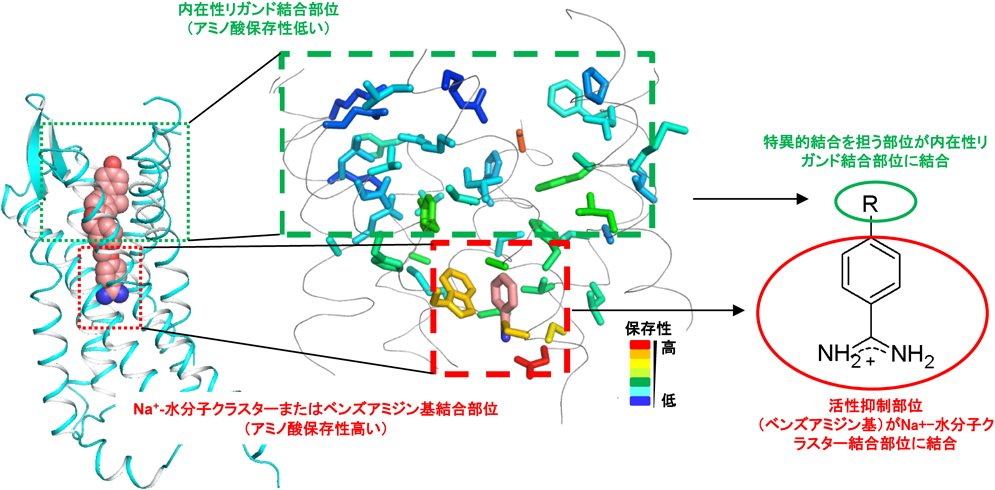
ロイコトリエンB4受容体(BLT1)の結晶構造解析から明らかになった,ナトリウムイオン–水分子クラスターの機能を模倣する低分子化合物による不活性状態のGPCRの立体構造の安定化機構
理化学研究所横山特別研究室RIKEN Yokoyama Laboratory ◇ 〒230–0045 横浜市鶴見区末広町1–7–22 ◇ 1–7–22 Suehiro-cho, Tsurumi-ku, Yokohama 230–0045, Japan
発行日:2019年6月25日Published: June 25, 2019