がん細胞の薬剤耐性機構Molecular mechanisms of drug resistance in tumor cells
東京薬科大学Tokyo University of Pharmacy and Life Sciences ◇ 〒192–0392 東京都八王子市堀之内1432–1 ◇ 1432–1 Horinouchi, Hachioji-city, Tokyo 192–0392, Japan
発行日:2019年8月25日Published: August 25, 2019
分子標的薬の開発によりがん治療に進展がみられているが,薬剤に対する耐性が生じ,がんの再増殖が起こることが治療の問題点である.薬剤耐性に関する研究は盛んに行われており,STAT3(signal transducer and activator of transcription 3)やFAK(focal adhesion kinase)などの分子の活性化によるシグナル伝達が薬剤耐性を亢進させていることが示されている.しかし,特異性の問題から,これらのシグナル伝達系を阻害するための優れた標的分子は少ない.我々はスクリーニングにより,さまざまながんで高発現するZIC5ががん細胞の内因的な薬剤耐性および薬剤に曝露されてから獲得する獲得耐性に寄与しており,ZIC5を阻害することで「がん細胞の薬剤耐性」を制御できることを明らかにした.
© 2019 公益社団法人日本生化学会© 2019 The Japanese Biochemical Society
分子標的薬の出現により特定のタンパク質やシグナル伝達系を阻害することが可能となりがん治療の効果を高めているが,薬剤耐性細胞の出現によりその治療効果は限定的になる.多くのがんは,がん原遺伝子やがん抑制遺伝子に変異や増幅などの異常が起き,細胞内のシグナル伝達経路が異常になることで生じる.約半数のメラノーマ症例において,BRAF(B-Raf proto-oncogene, serine/threonine kinase)というRAFファミリーに属するセリントレオニンキナーゼをコードする遺伝子に変異が入っており,異常に活性化したBRAFが「がん化」の要因であることが明らかにされている.メラノーマに対する分子標的薬として,変異型BRAFを抑制する低分子化合物が開発されており,このBRAF阻害薬は,BRAF変異を持つ転移性メラノーマ症例に対して高い奏効率を示し,全生存期間や無増悪生存期間を有意に延ばした1).しかし,半年ほど経つと,BRAF阻害薬に対する薬剤耐性を示すメラノーマが再び増殖を始めてしまう.このような現象は多くのがん治療においてみられ,がん治療における課題となっている.薬剤耐性が生じる分子機構についてのこれまでの知見と,我々が発見した新たな分子機構と治療標的について本稿で紹介する.
薬剤耐性機構の研究はさまざまながん種において研究されており,数多くの研究報告がある.本稿ではメラノーマにおける薬剤耐性機構を中心に話を進めていく.一般的に,がん細胞に薬剤耐性が生じるには,治療初期にがん細胞が死滅しないこと,そして生き残った細胞に変化が生じ,薬剤の存在下でも増殖可能になることが必要である(図1A).それぞれの分子機構について報告されていることを以下に記した.
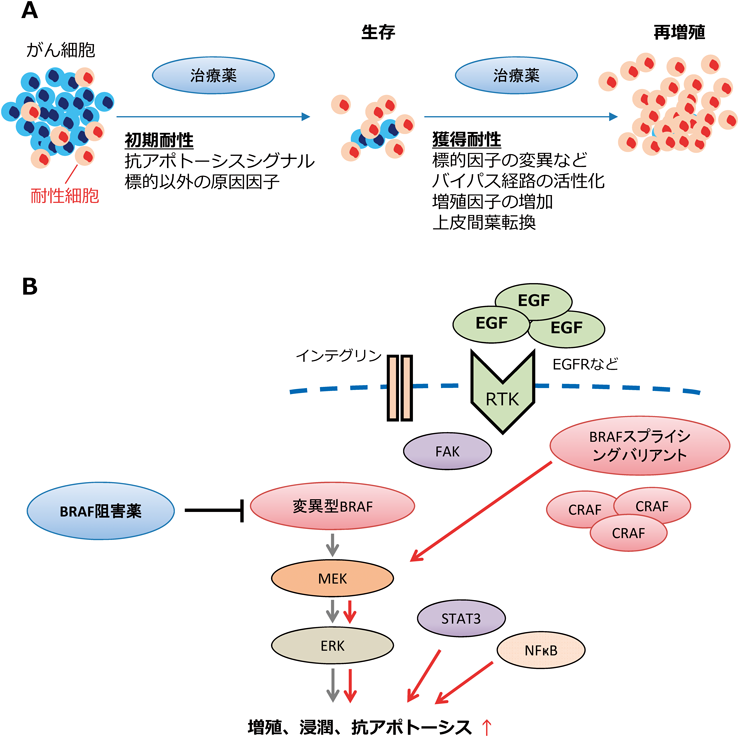
(A)治療薬がはじめは奏功するがん集団の中に,がん細胞の内因的要因により細胞死を回避する集団が存在し,残存する(初期耐性).残存したがん細胞は治療薬の存在下においても増殖できるようになり(獲得耐性),再びがんが大きくなる.(B)BRAF活性化変異が要因であるメラノーマに対して,BRAF阻害薬が奏功するが,さまざまなメカニズムにより薬剤耐性が生まれる.略称については本文参照.
「治療初期にがん細胞が死滅しない」現象に深く関わるのは,抗アポトーシスシグナルである.メラノーマの約半数の症例において,BRAFが異常活性化していることを先述したが,このBRAFは,各種RAFタンパク質→MEK(meiosis-specific serine/threonine-protein kinase)→ERK(extracellular signal-regulated kinase)という一連のキナーゼタンパク質を順に活性化する経路であるMAPK(mitogen-activated protein kinase)経路を活性化する.リン酸化を受け活性化したERKタンパク質は,抗アポトーシス因子であるMcl-1(myeloid cell leukemia 1),Bcl-2(B-cell leukemia/lymphoma 2),Bcl-XL(B-cell lymphoma extra-large)の発現を促進する2).Mcl-1は,Bclファミリーに属するタンパク質をコードしており,アポトーシスを阻害し細胞生存を促進する.Bcl-2は,ミトコンドリア外膜に局在するタンパク質をコードしており,アポトーシスを阻害する.Bcl-XLもBCL2ファミリーに属するタンパク質をコードしており,スプライシングバリアントのロングフォームである.このロングフォームであるBcl-XLはアポトーシスに対して阻害的に働く3).したがって,BRAF阻害薬により,これらの抗アポトーシス因子の発現が減少し,細胞死が誘導される.しかし,これらの抗アポトーシス因子はMAPK経路だけでなく,NFκB(nuclear factor kappa B)やSTAT3(signal transducer and activator of transcription 3)によっても発現誘導される4).
興味深いことに,EGFR(epidermal growth factor receptor)→RAS(rat sarcoma viral oncogene homolog)→RAF→MEK→ERKの一連の増殖シグナルの異常活性化が主要因であるがん細胞において,この経路を遮断する分子標的薬はSTAT3の活性化を引き起こし,抗アポトーシス因子の発現を促すことで薬剤耐性が生じることが報告されている5).この報告は肺がん細胞や大腸がん細胞を用いて示されているが,我々がBRAF活性化変異を持つメラノーマ細胞を用いて検証したところ,確かにBRAF阻害薬によりSTAT3の活性化の指標であるリン酸化STAT3が増加していた6).したがって,メラノーマ治療において,BRAF阻害薬によりBRAF→MEK→ERK経路を遮断して抗アポトーシス(生存促進)シグナルを阻害しても,STAT3が活性化することで再び抗アポトーシス因子の発現が誘導され,アポトーシスが回避されることになる.このようなメカニズムにより,一部のがんは分子標的薬の存在下でも生き残り,さらに下記のメカニズムを獲得したがん細胞は再び増殖を開始する.
「薬剤の存在下で再びがん細胞が増殖する現象」には,増殖シグナルの再活性化が関与する.BRAF阻害薬により阻害されるBRAF→MEK→ERK経路は,抗アポトーシス因子の発現を誘導するだけでなく,増殖促進シグナルとして働いている.BRAF阻害薬は変異型BRAFに起因するERKの異常活性化を阻害することで効果を発揮するが,BRAF阻害薬の存在下でも,ERKを活性化する機構が生じてしまうと,メラノーマは再び増殖を始めてしまう.ERKを再活性化しうる分子機構については数多く報告されており,以下に列挙した(図1B).
2011年のPoulikakosらの報告7)によると,BRAF阻害薬に対する耐性を持ったメラノーマ細胞は,変異型BRAFのスプライシングバリアントを発現しており,このタンパク質[p61BRAF(V600E)]が耐性に寄与していることを明らかにしている.p61BRAF(V600E)は,RAS結合ドメインを欠いており,RASの活性化なしに二量体を形成できる.このタンパク質を発現している細胞はRAF阻害薬の存在下でもERKが活性化する.実際にBRAF阻害薬に対して耐性を示した19症例のうち,6症例においてRAS結合ドメインを欠いたBRAFスプライシングバリアントの発現が認められていることから,BRAF遺伝子のスプライシングバリアントの発現がBRAF阻害薬に対する耐性機構の一つであることが示されている7).
MAPK経路により活性化されるERKタンパク質は,MAPK経路の構成因子であるEGFRやSOS(SOS Ras/Rac guanine nucleotide exchange factor),RAFをリン酸化することで,受容体型チロシンキナーゼを介するMAPK経路にネガティブフィードバックをかける.また,活性化したERKは,MAPK経路に対して抑制的に働くSproutyメンバータンパク質やDUSPファミリータンパク質の発現を誘導する8).BRAFやKRASなどのMAPK経路構成因子に活性化変異が入っているがん細胞においては,このネガティブフィードバック機構によりEGFRからのシグナルが遮断されても,変異タンパク質により異常にシグナルが活性化し続ける.このようながん細胞に対して,BRAFまたはKRAS阻害薬を添加すると,変異タンパク質からのシグナル活性化は抑制され,これまで発動し続けていたネガティブフィードバック機構がなくなる.したがって,受容体を介するシグナルが細胞内に伝達されやすくなる.このとき,細胞外に受容体型チロシンキナーゼやRASを活性化するリガンド分子が多く分泌されていれば,再びMAPK経路は活性化を受ける.
上記のように,変異型BRAFやKRASを阻害した細胞においては,受容体型チロシンキナーゼ(receptor tyrosine kinase:RTK)を介するシグナル伝達が活性化されやすくなる.RTKは増殖因子や成長因子と結合,活性化し,細胞内でRAS→RAF→MEK→ERKの一連のシグナル伝達経路などを活性化することで細胞増殖を促す.BRAF阻害薬に対する耐性を獲得したメラノーマ細胞株において,EGFやEGFRの発現が亢進していることが報告されている9).さらに,CRAF(Raf-1 proto-oncogene, serine/threonine kinase)量の増大やAKT(AKT serine/threonine kinase)活性の亢進,PDGFRβ(platelet derived growth factor receptor beta)量の増加,NRAS(NRAS proto-oncogene, GTPase)変異もBRAF阻害薬耐性と関連している場合がある10, 11).また,肺がん細胞や乳がん細胞においてRTK阻害薬を添加すると,FGF(fibroblast growth factor)やIL6(interleukin 6)の発現が亢進し,FGFシグナルやIL受容体からのシグナル伝達経路が活性化し,それによるSTAT3の活性化により抗アポトーシスシグナルが活性化されることが報告されている5).
2015年のHirataらの報告12)によると,BRAF阻害薬の投与によりメラノーマ付随線維芽細胞から細胞外マトリックスが異常に分泌され,近傍のメラノーマ細胞はインテグリンを介したシグナル伝達系の活性化を受ける.細胞外マトリックスと結合したインテグリンはFAK(focal adhesion kinase)とSrc(SRC proto-oncogene, non-receptor tyrosine kinase)を活性化し,これらはさらにRAS→RAF→MEK→ERK経路を活性化する.この分子機構の働きにより,BRAF阻害薬の存在下でもERKが再活性化してしまい,メラノーマは再び増殖を始める12).
上皮系の性質を持つ細胞が間葉系の性質を持つ細胞へと変化する現象である上皮間葉転換(epithelial-mesenchymal transition:EMT)は,さまざまな発生段階において見いだされた.初期胚における原腸陥入において,初期胚の上皮に該当する外胚葉組織がEMTを起こし,間葉系の性質に変化し,外胚葉から侵入して中胚葉や内胚葉組織を形成する.さらに,神経冠は神経外胚葉組織である神経管の一部からEMTを起こしてさまざまな場所に移動し,メラノサイト,末梢神経,グリア細胞,結合組織などに分化する13).
正常組織発生においてのみならず,がん組織内においてもさまざまなシグナルによってEMTが誘導される.EMTを誘導するシグナルとして,TGFβ(transforming growth factor beta)シグナル,Wntシグナル,インテグリンシグナルなどが知られている.がん組織内において,がんの浸潤先端などの一部の部位でEMTは誘導され,がん不均一性の要因となる.EMTが誘導される際には,Snail(snail family transcriptional repressor),ZEB(zinc finger E-box binding homeobox),Twist(twist family bHLH transcription factor)などの転写因子が活性化され,これらが上皮性遺伝子の発現を抑制し,間葉系遺伝子の発現を誘導する14).がん進展におけるEMTの関与は20年以上前に提唱され,転移がん組織のマイクロアレイ解析により,がん転移の際に多くのEMT関連因子が発現変動していることや,EMT関連転写因子により直接発現抑制を受けるEカドヘリンの発現低下と予後悪化との関連が示唆されてきた15).しかし,最近では,EMTプログラムが部分的に引き起こされた状態ががんの進展により深く関与していることが明らかにされている.膵がんモデルマウスにおいてSnailやTwistを欠損させてもコントロールマウスと有意差なく転移を形成すること16)や,間葉系マーカー遺伝子であるS100A4(S100 calcium binding protein A4)やビメンチンの発現が誘導されていない乳がん細胞が主に転移していることが報告され17),EMTと転移との関連に疑問が投げかけられた.2018年にPastushenkoらは,がん転移において上皮と間葉だけでなく,その中間の性質を持つがん細胞が出現していることを明らかにした18).彼らは,表皮扁平上皮がんモデルマウス(毛包細胞において活性化型KRasを発現させ,p53を欠損させたマウス)や乳がんモデルマウスを使用し,派生したがん細胞の性質をさまざまな細胞表面マーカーを用いて調査した.その結果,上皮系細胞間接着因子であるEpCAM(epithelial cell adhesion molecule)の発現を失っているがん細胞において,他のマーカー発現がヘテロな集団を形成していることが明らかになった.具体的には,CD51, CD61, CD106などの細胞表面タンパク質の発現がそれぞれ異なる複数の集団に分類された.CD51, CD61, CD106の発現がすべてない細胞集団(トリプルネガティブ)に対して,CD51, CD61, CD106の発現がすべてある細胞集団(トリプルポジティブ)の方が間葉性遺伝子群の発現が多く,上皮性遺伝子群の発現が低い.これらの細胞集団の性質を調べたところ,トリプルポジティブの細胞集団ほどin vitroにおける浸潤能が高いことが明らかになった.しかし,これらの細胞群をマウスの静脈内に投与し,肺への転移能を調べたところ,トリプルネガティブな細胞群で転移が顕著に多く観察されている.したがって,完全に間葉系の性質を獲得したがん細胞よりも,上皮間葉転換の中間体がより転移を起こすことが示唆されている18).
EMTプログラムによりがん細胞の転移だけでなく,幹細胞性も促進される19).これは,2008年にManiらが,乳腺上皮細胞に対してSnailの導入やTGFβ処理によりEMTを誘導したところ,スフィア形成(幹細胞性を試験する手法の一つで,足場のない状態での増殖をアッセイする方法)が亢進することや幹細胞マーカータンパク質の発現が高くなることを示した実験により明らかになった19).
さらに,幹細胞性質が亢進すると,薬剤やガンマ線照射などの治療に対しての抵抗性も増す.これは培養細胞系において主に明らかにされていたが,近年,EMTをトレースできるin vivoモデルにおいてもEMTと薬剤耐性の関係が明らかにされている.一度間葉に転換したがん細胞においてRFP(red fluorescent protein)が消失しGFP(green fluorescent protein)が発現するようなシステムを導入した乳がんモデルマウスを用いて,肺転移に完全なEMTは必要でないことが示されたが,このとき,抗がん剤耐性にEMTが関与するかについても試験されている.この乳がんモデルマウスにCTX(cyclophosphamide;抗がん剤の一種)を投与すると,肺転移巣に存在するがん細胞はほとんどがGFPポジティブな細胞であった.すなわち,EMTを起こした細胞が抗がん剤存在下でも生き残り,転移を起こしたと考えられる17).また,SnailやTwistを欠損させた膵がんモデルマウスにおいても,SnailやTwist欠損によるがん転移の抑制はみられなかったが,ゲムシタビン(抗がん剤の一種)に対する感受性が亢進することが示されている16).これらのことから,EMTはがん細胞の薬剤耐性(抵抗性)を促進する分子機構であることが示されている.
物質の輸送を行うタンパク質である膜貫通型タンパク質トランスポーターの一種であるABCトランスポーターは分子内によく保存されたATP結合領域を持ち,ATPの加水分解を利用して物質の輸送を行う.薬の排出にも関与していることから,ABCB1はさまざまながんで薬剤耐性に関与していることが報告されている.メラノーマ細胞においては,ABCB5タンパク質が幹細胞性の高い細胞に多く発現し,薬剤耐性に関与していることが明らかにされている20).
EMTプログラムの亢進がメラノーマの進展に関与していることから21),我々はEMTにおいて発現低下する上皮性細胞間接着因子であるEカドヘリンの発現を指標として,メラノーマ増悪に関与するタンパク質のスクリーニングを行った6).メラノサイトはEカドヘリンを発現しており,Eカドヘリンを介してケラチノサイトと相互作用し増殖が抑制される.メラノサイトががん化しメラノーマが進展するにつれて,Eカドヘリンやその他のメラノサイト分化マーカータンパク質の発現は低下する.また,メラノサイトががん化すると,メラノサイト前駆細胞の性質を持つことが知られている22).これは,ゼブラフィッシュのBRAF遺伝子に活性化変異を導入し,同時にp53遺伝子を欠失させたトランスジェニックゼブラフィッシュを使用した研究により明らかにされた.このトランスジェニックゼブラフィッシュに発生したメラノーマが神経冠前駆細胞に類似した遺伝子発現を示すことを,トランスクリプトーム解析により明らかにしている.
このように,メラノサイトは神経冠細胞が分化して生じること,メラノーマは神経冠前駆細胞に類似すること,神経冠細胞の形成にはEMTが重要であることなどから,我々は神経冠細胞の形成に重要な働きをする遺伝子の中にメラノーマ増悪に関与する遺伝子が多く存在しているのではないかと推測した.神経冠形成に必須の遺伝子群を抜粋し,メラノーマの悪性形質を促進する遺伝子をスクリーニングにより探索した.神経冠形成に必須の遺伝子群は,アフリカツメガエルの初期胚にアンチセンスモルフォリノオリゴを注入し,遺伝子発現を抑制する実験において数多く同定されている.神経冠形成に必須であることが示されている遺伝子のうち,がんにおける役割が未知な遺伝子群に対してsiRNA(small interfering RNA)ライブラリーを作製し,siRNAをメラノーマ細胞に導入後,免疫染色によりEカドヘリン量を定量する方法でスクリーニングを行った(図2).その結果,六つの遺伝子が同定された.我々はその中から,ヒト正常組織での発現がきわめて限定的であったZIC5(Zic family member 5)に着目し,ZIC5がメラノーマ進展に与える影響について研究を進めてきた(図3A).
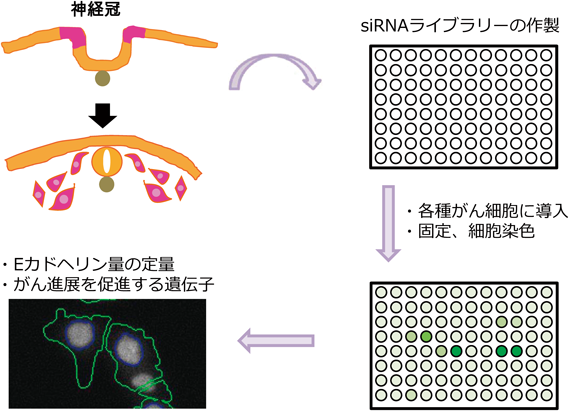
神経冠形成に必須の遺伝子群に対してsiRNAライブラリーを作製し,各種がん細胞に導入した.細胞を固定し,Eカドヘリンに対する抗体を用いて細胞染色を行い,In Cell Analyzerを使用してEカドヘリン量を定量した.Eカドヘリン発現に対して抑制的に機能している遺伝子をピックアップし,がん進展に関与している遺伝子を同定した.
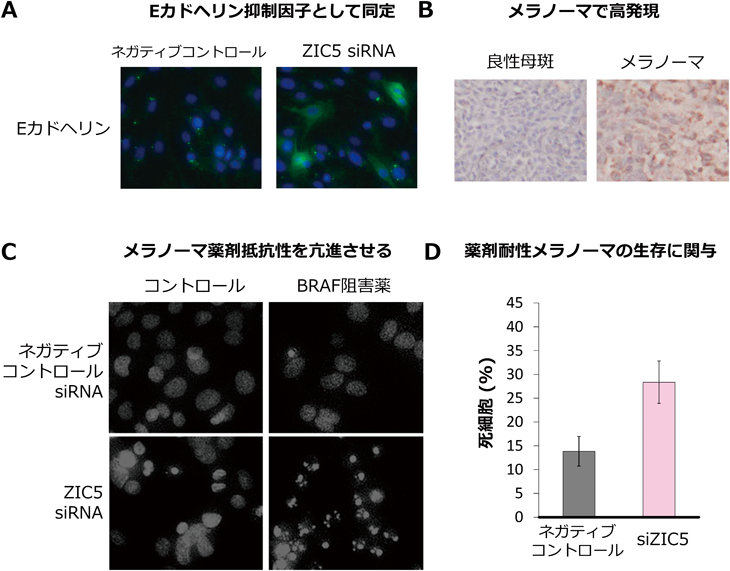
(A)ZIC5を発現抑制すると,Eカドヘリン発現が回復した.(B)ヒトメラノーマ組織でZIC5は高発現した(茶色).(C)ZIC5を発現抑制してからBRAF阻害薬を添加することで,ほとんどのメラノーマ細胞がアポトーシスの兆候である核の断片化を起こした.(D)BRAF阻害薬耐性メラノーマ細胞においてZIC5を発現抑制すると,細胞死が引き起こされた.
実際に,ヒトのメラノーマ組織でZIC5の発現が亢進しているかについてZIC5抗体を用いた免疫染色を行ったところ,約70%の症例でZIC5の高発現が認められた.さらに,ステージの高いメラノーマにおいてより多くZIC5が存在していることが示唆された(図3B).各種メラノーマ細胞株におけるZIC5の発現量を比較したところ,分化度の低い細胞株ほどZIC5の発現が高いことが明らかとなった.さらに,メラノーマ細胞においてZIC5を発現抑制すると,メラノーマ細胞の増殖,運動,浸潤が抑制されること,免疫不全マウスの皮下におけるメラノーマ細胞の増殖や肺転移が抑制されることを明らかにした6).
薬剤耐性に対する影響を調べたところ,驚くべきことに,ZIC5を発現抑制してからBRAF阻害薬を添加したメラノーマ細胞は,ほとんどの細胞がアポトーシスを起こした(図3C).通常,BRAF阻害薬の存在下で長期的にメラノーマ細胞を培養していると,そのうちにBRAF阻害薬の存在下でも増殖可能な薬剤耐性細胞が出現し,メラノーマ細胞は再び増殖を始める.しかし,ZIC5を発現抑制したメラノーマ細胞ではBRAF阻害薬でほとんどすべての細胞が死滅するため,そのような耐性細胞は現れなかった.さらに,すでにBRAF阻害薬に対して耐性を獲得した耐性株においてもZIC5の阻害により細胞死が誘導された(図3D).これらのことから,ZIC5はメラノーマ増悪を促進させる分子であり,その抑制がメラノーマの抑制,特に薬剤耐性を抑制することが明らかとなった6).
ZIC5はEカドヘリン発現を抑制する遺伝子としてスクリーニングにより同定された.我々は,実際にZIC5がEカドヘリンプロモーター活性を抑制すること,Eカドヘリン転写開始点上流領域に結合することを明らかにしている.しかし,Eカドヘリンの発現がまったくなく,ZIC5を発現抑制してもEカドヘリンの発現が回復しないメラノーマ細胞株においても,ZIC5の発現抑制は増殖低下や細胞運動の低下,抗がん剤に対する抵抗性の低下を引き起こした.したがって,Eカドヘリンの発現制御以外のメカニズムによりZIC5がメラノーマの悪性形質を制御していることが示唆された.ZIC5がどのような遺伝子の発現を変動させてメラノーマの薬剤耐性を促進しているかを明らかにするため,ZIC5の発現抑制により変動するトランスクリプトーム解析を行った.変動遺伝子に対するパスウェイ解析の結果,focal adhesion(接着斑)に関連する遺伝子が多く発現変動していることが明らかになった.focal adhesionにおいて活性化するFAKはメラノーマの薬剤耐性を促進する分子であることが知られていたことから,ZIC5とFAKの活性化との関連を検証した.ZIC5を発現抑制した際にFAKの活性化の指標であるリン酸化FAK量に変動がみられるかを検証したところ,ZIC5を発現抑制するとリン酸化FAK量が顕著に減少することが明らかになった.ZIC5によるFAKの活性化には,ZIC5により発現誘導を受けるPDGFD(platelet derived growth factor D)が関与していることも明らかになった.PDGFDは,血小板由来成長因子の一種であり,チロシンキナーゼ型PDGF受容体を介したシグナル伝達経路を活性化することが知られていた.活性化したPDGF受容体に結合し活性化するSrcタンパク質は,FAKと相互に活性を増強させることが報告されている.また,SrcはSTAT3を活性化することも報告されていたことから,ZIC5によりSTAT3の活性化に変動がある可能性が考えられた.先述したように,STAT3は抗アポトーシス因子の発現を誘導する重要な薬剤耐性関連タンパク質である.ZIC5を発現抑制すると,メラノーマ細胞におけるSTAT3の活性化の指標であるリン酸化は著しく減少した.この減少はBRAF阻害薬の添加後により顕著にみられた.したがって,BRAF阻害薬を添加し,先述したMAPK経路のネガティブフィードバック機構を解いた状態の細胞において,ZIC5→PDGFD→FAK/Src→STAT3の活性化経路がより強く引き起こされると考えられる.そして,STAT3により抗アポトーシス経路が活性化し,薬剤耐性を亢進させると考えられる(図4).さらに,STAT3の活性を阻害した際やPDGFDの発現を抑制した際にZIC5の発現量も低下することから,これらのシグナル経路はポジティブフィードバックループを形成していることが明らかとなった.
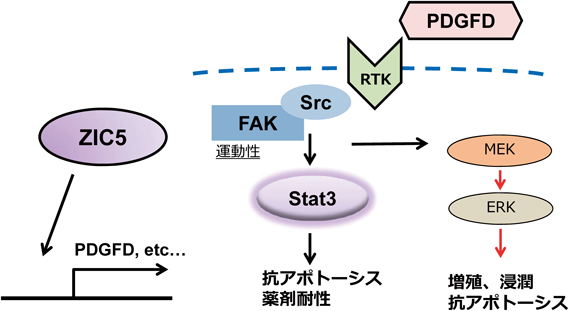
ZIC5はPDGFDの発現を誘導し,PDGFDはFAKやSTAT3を活性化する.FAKとSrcは相互に活性化し,MAPK経路の再活性化に寄与する.
すでにBRAF阻害薬に対する耐性を獲得している耐性株においてZIC5の作用機序を解析したところ,BRAF阻害薬で阻害したMAPK経路の再活性化に関与していることが示唆された.先述したように,FAKはRAS→RAF→MEK→ERK経路を活性化するため,ZIC5/PDGFDにより活性化したFAKはBRAF阻害薬で阻害した経路の再活性化を促すことができる.耐性株においてBRAF阻害薬とZIC5発現抑制を同時に起こすと,ERKのリン酸化が減少した.したがって,ZIC5が促進するシグナル経路がMAPK経路のバイパスシグナルとして働いていると考えられる(図4).
以上のことから,メラノーマ細胞において,ZIC5→PDGFD→FAK→STAT3→ZIC5のポジティブフィードバックループを形成しているシグナル経路に関わるどの因子を阻害しても,抗アポトーシスシグナルとMAPK経路に対するバイパス経路の活性化を阻害することができ,薬剤耐性を減弱させることができると考えられる.STAT3やFAKはすでに多くのがんの増悪に関与することが知られており,10年以上前から阻害薬の開発が試みられている.STAT3の阻害薬としては,STAT3の二量体化を阻害するS3I-20123)やS3I-175724),STAT3のDNA結合能を阻害するIS3 29525)など数多くの化合物が同定されているが,いまだに米国食品医薬品局(FDA)に承認されているものはない.その理由として,STAT3阻害薬は比較的細胞毒性が高いことがあげられる.臨床試験において,STAT3阻害薬はしばしば,倦怠感,下痢,感染症,末梢神経障害などの有害事象を引き起こすことが報告されている26).これらのSTAT3阻害薬による有害事象は,STAT3の生理機能を正常細胞において阻害したことに起因すると考えられる.STAT3のコンディショナルノックアウトマウスを使用した研究において,STAT3をノックアウトすると急性の腸炎および体重減少が引き起こされることが明らかにされている27).また,STAT3は造血細胞の分化に重要な役割を担っており,STAT3を骨髄細胞特異的に欠損させたマウスにおいて,骨髄系細胞の自律的な増殖や自然免疫の異常活性化が観察されている28).
FAKの阻害薬としては,PF-00562271, PF-04554878, VS-4718, GSK2256098, BI853520など多くの化合物が開発され臨床試験が行われている29).しかし,FAKも多くの正常組織で発現しており,造骨細胞の分化に必要であることが報告されていることから,FAKの阻害による有害事象が引き起こされる可能性が考えられる30).したがって,STAT3やFAKは多くのがんの増悪や薬剤耐性に関与していることが明らかになっているが,治療標的としての特異性は高くない.
ZIC5は正常ヒト組織における発現が非常に少ない.各種ヒト正常組織に対するRNAシークエンス解析のデータベースをみると,ZIC5は精巣と大脳皮質でのみ発現が検出されており,その他の組織では発現が確認されていないことがわかる.したがって,ZIC5は非常にがん特異性が高いタンパク質であるといえる.また,ZIC5により発現誘導を受けFAKやSTAT3の活性化を促進するPDGFDは,各種正常組織での発現がみられるが,PDGFDのノックアウトマウスはわずかに心臓の血管に異常がみられるものの,ほぼ正常であることが報告されている31).したがって,PDGFDを阻害しても正常組織に対する影響は少ないことが予想される.これらのことから,我々が同定したZIC5とPDGFDはがん細胞の薬剤耐性を減弱させるための治療標的として有望であると考えられる.
ここまで,メラノーマにおけるZIC5の役割について述べてきたが,ZIC5の他のがん種における役割についても明らかになっている.TCG A(The Cancer Genome Atlas)データベースでは,ZIC5はメラノーマのみならず,膀胱がん,大腸がん,食道がん,頭頸部扁平上皮がん,肝がん,肺腺がん,肺小細胞がん,前立腺がん,胃がん,食道がん,胸腺腫,子宮体がんで高発現していることが示唆されている.大腸がん,前立腺がん,非小細胞肺がん,肝がん,グリオーマにおいては培養細胞レベルでZIC5の機能解析が行われている(図5).
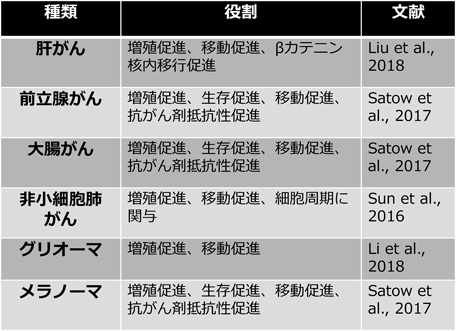
これまで報告されているZIC5の役割を表にまとめた.
我々は,大腸がんと前立腺がん細胞株を用いて,ZIC5の機能解析を行った.これらのがん細胞においてZIC5を発現抑制すると,PDGFDの発現が低下し,FAKとSTAT3の活性化の指標であるリン酸化量が減少した.また,ZIC5やPDGFDを発現抑制してから抗がん剤を添加することで,細胞死誘導率が亢進することが明らかになった.これらのことから,大腸がん細胞や前立腺がん細胞においてもZIC5がPDGFDの発現を誘導し,FAK, STAT3の活性化に寄与し,抗がん剤に対する抵抗性を亢進させているものと考えられる.しかし,STAT3の発現がない前立腺がん細胞においても,ZIC5の発現抑制により細胞死が亢進したことから,ZIC5はSTAT3を介さない経路でも抗がん剤抵抗性を亢進させることが示唆された.ZIC5が制御する他のがん関連因子を抗体アレイを使用して探索した結果,リン酸化サイクリン依存性キナーゼ阻害因子(p27kip1)はZIC5により負の制御を受け,HSP60(heat shock protein family D)は正の制御を受けることが明らかになった32).P27kipはリン酸化されることで安定化し,前立腺がんの進展に対して抑制的に作用することが知られている33).また,HSP60はがん細胞の生存に対して促進的に作用するタンパク質であることが知られている34).これらのタンパク質はPDGFDの発現抑制により変動しなかったことから,ZIC5がPDGFDとは無関係に制御しており,がん細胞の生存や進展に関与していると考えられる.
Sunらの報告35)によると,非小細胞肺がん細胞でZIC5を発現抑制すると,細胞増殖や細胞移動が抑制され,細胞周期のG2停止が引き起こされる.また,ZIC5の発現抑制により,CDC25C(cell division cycle 25C),リン酸化CDK1(cyclin dependent kinase 1),CCNB1(cyclin B1)の減少が観察されている35).Liuらは,肝細胞がんにおいてZIC5の機能解析を行っており,ZIC5の発現抑制により,細胞増殖,細胞移動や免疫不全マウスの皮下における造腫瘍能が低下することを示している36).また,ZIC5の過剰発現によりβカテニンの核内移行が促進されることを示している36).これらの知見より,ZIC5はさまざまな種類のがんにおいて,増殖や生存に促進的な役割を持つタンパク質であると考えられる.
我々は,PDGFDがメラノーマ,大腸がん,前立腺がん細胞の悪性形質(増殖,移動,生存など)を促進するタンパク質であることを明らかにしているが,これまでにさまざまながん種においてPDGFDの役割が報告されている.腎細胞がんで高発現し,増殖や転移を促進すること37),乳がん細胞においてビメンチンやZEB-2などのEMT関連因子の発現を誘導し,増殖や生存を正に制御すること38),卵巣がんで高発現し,MMP(matrix metalloproteinase)を誘導し,浸潤,転移を促進すること39),ゲムシタビン耐性を持つ肝細胞がんで高発現し,EMT誘導を正に制御していること40),子宮内膜がん細胞の悪性形質を亢進させること41),舌がんにおいて,p38, AKT, ERKの活性化を介して悪性形質を促進すること42),などさまざまながん種において一貫してがん進展を促進する役割が報告されている.
Liらは,グリオーマで発現低下がみられるマイクロRNAの一つであるmiR-761が,ZIC5 mRNAの3′ UTRに結合してZIC5量を低下させていることを報告している43).グリオーマ細胞でZIC5を発現抑制すると,細胞増殖や細胞移動が低下することを示しており,グリオーマにおけるmiR-761の発現低下がZIC5の発現増加を介してグリオーマの増悪を促進していることを示唆している43).
我々は,ZIC5が核内に局在し,転写因子として機能することを明らかにした6).しかし,ZIC5がどのように核内に移行するかは不明である.他のZICファミリータンパク質であるZIC2はI-mfa(MyoD family inhibitor)というタンパク質と結合すると細胞質に移行し,転写活性がなくなること44),ZIC3はその核内移行にSUMO化を受けることが重要であることが示されており45),ZICタンパク質の活性を制御するさまざまな分子メカニズムがあることを示唆している.ZIC5の活性がどのように制御されているかはまだわかっていないが,我々はZIC5結合タンパク質のスクリーニングを行い,いくつかのZIC5結合タンパク質を同定している.この中からZIC5の機能に重要なものを探索することで,ZIC5の制御機構が明らかになると期待している.
現在我々は,ZIC5を指標として,がん細胞の薬剤耐性を阻害する化合物のスクリーニングを行っている.ZIC5が持つジンクフィンガードメインを欠損させた変異体は核内移行できず,STAT3のリン酸化も増加させないことから,ZIC5は核内で機能することががん細胞の薬剤耐性にとって重要だと考えられる.したがって,ZIC5の核内移行を阻害する化合物をスクリーニングにより探索している.また,ZIC5はmRNA量とタンパク質量が相関しないことが多く,ZIC5タンパク質量の調整には転写後の調節が深く関わっていると考えられる.したがって,ZIC5のタンパク質量を減少させる化合物も同時に探索している.PDGFDも治療標的として有望であり,その作用機序と阻害抗体の作製を進めているが,ZIC5はPDGFDを介さない経路でも薬剤耐性を促進していることが示唆されたため32),ZIC5,もしくはZIC5とPDGFDを両方阻害する戦略が多くのがん細胞にとって有効になると考えている.ZIC5やPDGFDを阻害した細胞に既存の抗がん剤や分子標的薬を添加すると,通常よりも低い濃度で高い細胞死誘導率を示す.そのため,ZIC5阻害薬およびPDGFD阻害抗体は既存の治療薬の細胞死誘導効率を高め,その使用量も少なくすることができる見込みがあり,非常に興味深い治療標的だと考えている.
低分子化合物の多くの標的は酵素活性を持つタンパク質であり,抗体医薬の標的は細胞表面受容体や分泌因子である.したがって,転写因子に対する創薬は簡単ではないと考えられるが,それにチャレンジし成功した場合には他の多くの転写因子に対する創薬を活性化することにもつながると考えている.近年,分子間相互作用解析装置や核酸医薬開発の進展がみられており,どのような分子でも治療標的になる可能性はある.より多くのがん生存戦略を明らかにし,それを阻害するための治療標的分子を多く同定しておく必要がある.
本稿で紹介した筆者の研究は,東京薬科大学生命科学部生命医科学科ゲノム病態医科学研究室で行ったものになります.ご指導とご助言をいただいた深見希代子教授に心より感謝申し上げます.また,この研究は研究室の多くの学生さんが進めてくれた成果になります.これまで研究に携わった方々や多くの共同研究者の方々にこの場を借りて深く感謝いたします.
本総説は2018年度奨励賞を受賞した.
1) Chapman, P.B., Hauschild, A., Robert, C., Haanen, J.B., Ascierto, P., Larkin, J., Dummer, R., Garbe, C., Testori, A., Maio, M., et al.; BRIM-3 Study Group. (2011) Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N. Engl. J. Med., 364, 2507–2516.
2) Boucher, M.J., Morisset, J., Vachon, P.H., Reed, J.C., Lainé, J., & Rivard, N. (2000) MEK/ERK signaling pathway regulates the expression of Bcl-2, Bcl-XL, and Mcl-1 and promotes survival of human pancreatic cancer cells. J. Cell. Biochem., 79, 355–369.
3) García-Sáez, A.J. (2012) The secrets of the Bcl-2 family. Cell Death Differ., 9, 1733–1740.
4) Lee, H., Herrmann, A., Deng, J.H., Kujawski, M., Niu, G., Li, Z., Forman, S., Jove, R., Pardoll, D.M., & Yu, H. (2009) Persistently activated Stat3 maintains constitutive NF-kappaB activity in tumors. Cancer Cell, 15, 283–293.
5) Lee, H.J., Zhuang, G., Cao, Y., Du, P., Kim, H.J., & Settleman, J. (2014) Drug resistance via feedback activation of Stat3 in oncogene-addicted cancer cells. Cancer Cell, 26, 207–221.
6) Satow, R., Nakamura, T., Kato, C., Endo, M., Tamura, M., Batori, R., Tomura, S., Murayama, Y., & Fukami, K. (2017) ZIC5 drives melanoma aggressiveness by PDGFD-mediated activation of FAK and STAT3. Cancer Res., 77, 366–377.
7) Poulikakos, P.I., Persaud, Y., Janakiraman, M., Kong, X., Ng, C., Moriceau, G., Shi, H., Atefi, M., Titz, B., Gabay, M.T., et al. (2011) RAF inhibitor resistance is mediated by dimerization of aberrantly spliced BRAF(V600E). Nature, 480, 387–390.
8) Lito, P., Pratilas, C.A., Joseph, E.W., Tadi, M., Halilovic, E., Zubrowski, M., Huang, A., Wong, W.L., Callahan, M.K., Merghoub, T., et al. (2012) Relief of profound feedback inhibition of mitogenic signaling by RAF inhibitors attenuates their activity in BRAFV600E melanomas. Cancer Cell, 22, 668–682.
9) Girotti, M.R., Pedersen, M., Sanchez-Laorden, B., Viros, A., Turajlic, S., Niculescu-Duvaz, D., Zambon, A., Sinclair, J., Hayes, A., Gore, M., et al. (2013) Inhibiting EGF receptor or SRC family kinase signaling overcomes BRAF inhibitor resistance in melanoma. Cancer Discov., 3, 158–167.
10) Su, F., Bradley, W.D., Wang, Q., Yang, H., Xu, L., Higgins, B., Kolinsky, K., Packman, K., Kim, M.J., Trunzer, K., et al. (2012) Resistance to selective BRAF inhibition can be mediated by modest upstream pathway activation. Cancer Res., 72, 969–978.
11) Nazarian, R., Shi, H., Wang, Q., Kong, X., Koya, R.C., Lee, H., Chen, Z., Lee, M.K., Attar, N., Sazegar, H., et al. (2010) Melanomas acquire resistance to B-RAF(V600E) inhibition by RTK or N-RAS upregulation. Nature, 468, 973–937.
12) Hirata, E., Girotti, M.R., Viros, A., Hooper, S., Spencer-Dene, B., Matsuda, M., Larkin, J., Marais, R., & Sahai, E. (2015) Intravital imaging reveals how BRAF inhibition generates drug-tolerant microenvironments with high integrin β1/FAK signaling. Cancer Cell, 27, 574–588.
13) Lim, J. & Thiery, J.P. (2012) Epithelial–mesenchymal transitions: Insights from development. Development, 139, 3471–3486.
14) Sleeman, J.P. & Thiery, J.P. (2011) SnapShot: The epithelial–mesenchymal transition. Cell, 145, 162.e1.
15) Alonso, S.R., Tracey, L., Ortiz, P., Pérez-Gómez, B., Palacios, J., Pollán, M., Linares, J., Serrano, S., Sáez-Castillo, A.I., Sánchez, L., et al. (2007) A high-throughput study in melanoma identifies epithelial-mesenchymal transition as a major determinant of metastasis. Cancer Res., 67, 3450–3460.
16) Zheng, X., Carstens, J.L., Kim, J., Scheible, M., Kaye, J., Sugimoto, H., Wu, C.C., LeBleu, V.S., & Kalluri, R. (2015) Epithelial-to-mesenchymal transition is dispensable for metastasis but induces chemoresistance in pancreatic cancer. Nature, 527, 525–530.
17) Fischer, K.R., Durrans, A., Lee, S., Sheng, J., Li, F., Wong, S.T., Choi, H., El Rayes, T., Ryu, S., Troeger, J., et al. (2015) Epithelial-to-mesenchymal transition is not required for lung metastasis but contributes to chemoresistance. Nature, 527, 472–476.
18) Pastushenko, I., Brisebarre, A., Sifrim, A., Fioramonti, M., Revenco, T., Boumahdi, S., Van Keymeulen, A., Brown, D., Moers, V., Lemaire, S., et al. (2018) Identification of the tumour transition states occurring during EMT. Nature, 556, 463–468.
19) Mani, S.A., Guo, W., Liao, M.J., Eaton, E.N., Ayyanan, A., Zhou, A.Y., Brooks, M., Reinhard, F., Zhang, C.C., Shipitsin, M., et al. (2008) The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell, 133, 704–715.
20) Frank, N.Y., Margaryan, A., Huang, Y., Schatton, T., Waaga-Gasser, A.M., Gasser, M., Sayegh, M.H., Sadee, W., & Frank, M.H. (2005) ABCB5-mediated doxorubicin transport and chemoresistance in human malignant melanoma. Cancer Res., 65, 4320–4333.
21) Alonso, S.R., Tracey, L., Ortiz, P., Pérez-Gómez, B., Palacios, J., Pollán, M., Linares, J., Serrano, S., Sáez-Castillo, A.I., Sánchez, L., et al. (2007) A high-throughput study in melanoma identifies epithelial–mesenchymal transition as a major determinant of metastasis. Cancer Res., 67, 3450–3460.
22) Kaufman, C.K., Mosimann, C., Fan, Z.P., Yang, S., Thomas, A.J., Ablain, J., Tan, J.L., Fogley, R.D., van Rooijen, E., Hagedorn, E.J., et al. (2016) A zebrafish melanoma model reveals emergence of neural crest identity during melanoma initiation. Science, 351, aad2197.
23) Siddiquee, K., Zhang, S., Guida, W.C., Blaskovich, M.A., Greedy, B., Lawrence, H.R., Yip, M.L., Jove, R., McLaughlin, M.M., Lawrence, N.J., et al. (2007) Selective chemical probe inhibitor of Stat3, identified through structure-based virtual screening, induces antitumor activity. Proc. Natl. Acad. Sci. USA, 104, 7391–7396.
24) Zhang, X., Sun, Y., Pireddu, R., Yang, H., Urlam, M.K., Lawrence, H.R., Guida, W.C., Lawrence, N.J., & Sebti, S.M. (2013) A novel inhibitor of STAT3 homodimerization selectively suppresses STAT3 activity and malignant transformation. Cancer Res., 73, 1922–1933.
25) Turkson, J., Zhang, S., Mora, L.B., Burns, A., Sebti, S., & Jove, R. (2005) A novel platinum compound inhibits constitutive Stat3 signaling and induces cell cycle arrest and apoptosis of malignant cells. J. Biol. Chem., 280, 32979–32988.
26) Beebe, J.D., Liu, J.Y., & Zhang, J.T. (2018) Two decades of research in discovery of anticancer drugs targeting STAT3, how close are we? Pharmacol. Ther., 191, 74–91.
27) Alonzi, T., Newton, I.P., Bryce, P.J., Di Carlo, E., Lattanzio, G., Tripodi, M., Musiani, P., & Poli, V. (2004) Induced somatic inactivation of STAT3 in mice triggers the development of a fulminant form of enterocolitis. Cytokine, 26, 45–56.
28) Welte, T., Zhang, S.S., Wang, T., Zhang, Z., Hesslein, D.G., Yin, Z., Kano, A., Iwamoto, Y., Li, E., Craft, J.E., et al. (2003) STAT3 deletion during hematopoiesis causes Crohn’s disease-like pathogenesis and lethality: A critical role of STAT3 in innate immunity. Proc. Natl. Acad. Sci. USA, 100, 1879–1884.
29) Lee, B.Y., Timpson, P., Horvath, L.G., & Daly, R.J. (2015) FAK signaling in human cancer as a target for therapeutics. Pharmacol. Ther., 146, 132–149.
30) Sun, C., Yuan, H., Wang, L., Wei, X., Williams, L., Krebsbach, P.H., Guan, J.L., & Liu, F. (2016) FAK promotes osteoblast progenitor cell proliferation and differentiation by enhancing wnt signaling. J. Bone Miner. Res., 31, 2227–2238.
31) Gladh, H., Folestad, E.B., Muhl, L., Ehnman, M., Tannenberg, P., Lawrence, A.L., Betsholtz, C., & Eriksson, U. (2016) Mice lacking platelet-derived growth factor D display a mild vascular phenotype. PLoS One, 11, e0152276.
32) Satow, R., Inagaki, S., Kato, C., Shimozawa, M., & Fukami, K. (2017) Identification of zinc finger protein of the cerebellum 5 as a survival factor of prostate and colorectal cancer cells. Cancer Sci., 108, 2405–2412.
33) Roy, S., Singh, R.P., Agarwal, C., Siriwardana, S., Sclafani, R., & Agarwal, R. (2008) Downregulation of both p21/Cip1 and p27/Kip1 produces a more aggressive prostate cancer phenotype. Cell Cycle, 7, 1828–1835.
34) Cornford, P.A., Dodson, A.R., Parsons, K.F., Desmond, A.D., Woolfenden, A., Fordham, M., Neoptolemos, J.P., Ke, Y., & Foster, C.S. (2000) Heat shock protein expression independently predicts clinical outcome in prostate cancer. Cancer Res., 60, 7099–7105.
35) Sun, Q., Shi, R., Wang, X., Li, D., Wu, H., & Ren, B. (2016) Overexpression of ZIC5 promotes proliferation in non-small cell lung cancer. Biochem. Biophys. Res. Commun., 479, 502–509.
36) Liu, L., Hu, X., Sun, D., Wu, Y., & Zhao, Z. (2018) ZIC5 facilitates the growth of hepatocellular carcinoma through activating Wnt/β-catenin pathway. Biochem. Biophys. Res. Commun., 503, 2173–2179.
37) Xu, L., Tong, R., Cochran, D.M., & Jain, R.K. (2005) Blocking platelet-derived growth factor-D/platelet-derived growth factor receptor beta signaling inhibits human renal cell carcinoma progression in an orthotopic mouse model. Cancer Res., 65, 5711–5719.
38) Ahmad, A., Wang, Z., Kong, D., Ali, R., Ali, S., Banerjee, S., & Sarkar, F.H. (2011) Platelet-derived growth factor-D contributes to aggressiveness of breast cancer cells by up-regulating Notch and NF-κB signaling pathways. Breast Cancer Res. Treat., 126, 15–25.
39) Wang, Y., Hu, C., Dong, R., Huang, X., & Qiu, H. (2011) Platelet-derived growth factor-D promotes ovarian cancer invasion by regulating matrix metalloproteinases 2 and 9. Asian Pac. J. Cancer Prev., 12, 3367–3370.
40) Wu, Q., Wang, R., Yang, Q., Hou, X., Chen, S., Hou, Y., Chen, C., Yang, Y., Miele, L., Sarkar, F.H., et al. (2013) Chemoresistance to gemcitabine in hepatoma cells induces epithelial-mesenchymal transition and involves activation of PDGF-D pathway. Oncotarget, 4, 1999–2009.
41) Wang, Y., Qiu, H., Hu, W., Li, S., & Yu, J. (2014) Over–expression of platelet-derived growth factor-D promotes tumor growth and invasion in endometrial cancer. Int. J. Mol. Sci., 15, 4780–4794.
42) Zhang, H., Sun, J.D., Yan, L.J., & Zhao, X.P. (2016) PDGF-D/PDGFRβ promotes tongue squamous carcinoma cell (TSCC) progression via activating p38/AKT/ERK/EMT signal pathway. Biochem. Biophys. Res. Commun., 478, 845–851.
43) Li, G.F., Li, L., Yao, Z.Q., & Zhuang, S.J. (2018) Hsa_circ_0007534/miR-761/ZIC5 regulatory loop modulates the proliferation and migration of glioma cells. Biochem. Biophys. Res. Commun., 499, 765–771.
44) Ishiguro, A., Inoue, T., Mikoshiba, K., & Aruga, J. (2004) Molecular properties of Zic4 and Zic5 proteins: Functional diversity within Zic family. Biochem. Biophys. Res. Commun., 324, 302–307.
45) Chen, L., Ma, Y., Qian, L., & Wang, J. (2013) Sumoylation regulates nuclear localization and function of zinc finger transcription factor ZIC3. Biochim. Biophys. Acta, 1833, 2725–2733.

東京薬科大学生命科学部講師.博士(理学).
2001年東京理科大学理工学部応用生物科学科卒業.03年東京大学大学院理学系研究科修士課程修了.06年同博士課程修了.同年 国立がんセンター研究所.11年東京薬科大学生命科学部助教.19年同講師.
研究テーマと抱負近年,様々な新しい治療法の出現によりがん治療効果は上昇しているが,根治しない症例も多くある.がん細胞は多様な性質を示し,治療戦略をすり抜けていく.がん細胞が生き残っていくメカニズムを様々な角度から明らかにし,より多くの治療標的を同定することが「がん根治」に繋がると考えている.
ウェブサイトhttp://toyaku-ls-genome.com/
趣味テニス,バドミントン.

This page was created on 2019-07-11T10:51:16.236+09:00
This page was last modified on 2019-08-08T11:32:34.000+09:00
このサイトは(株)国際文献社によって運用されています。