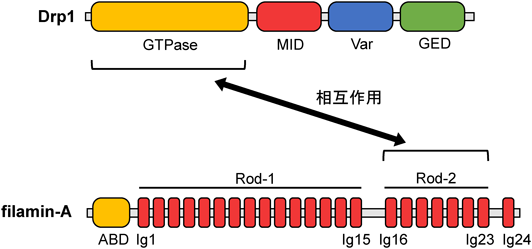
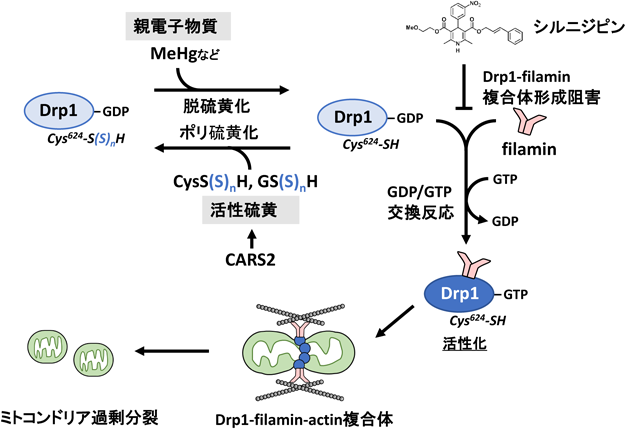
病態特異的なミトコンドリア–細胞骨格間相互作用を介した心筋の早期老化制御Regulation of cardiac early senescence via pathology-specific interaction between mitochondria and cytoskeleton
1 九州大学大学院薬学研究院創薬育薬研究施設統括室Department of Translational Pharmaceutical Sciences, Graduate School of Pharmaceutical Sciences, Kyushu University ◇ 〒812–8582 福岡県福岡市東区馬出3–1–1 ◇ 3–1–1 Maidashi, Higashi–ku, Fukuoka, Fukuoka 812–8582, Japan
2 自然科学研究機構生理学研究所(生命創成探究センター)心循環シグナル研究部門Division of Cardiocirculatory Signaling, National Institute for Physiological Sciences and Exploratory Research Center on Life and Living Systems, National Institutes of Natural Sciences ◇ 〒444–8787 愛知県岡崎市明大寺町字東山5–1 ◇ 5–1 Higashiyama, Myodaiji-cho, Okazaki, Aichi 444–8787, Japan
発行日:2019年8月25日Published: August 25, 2019