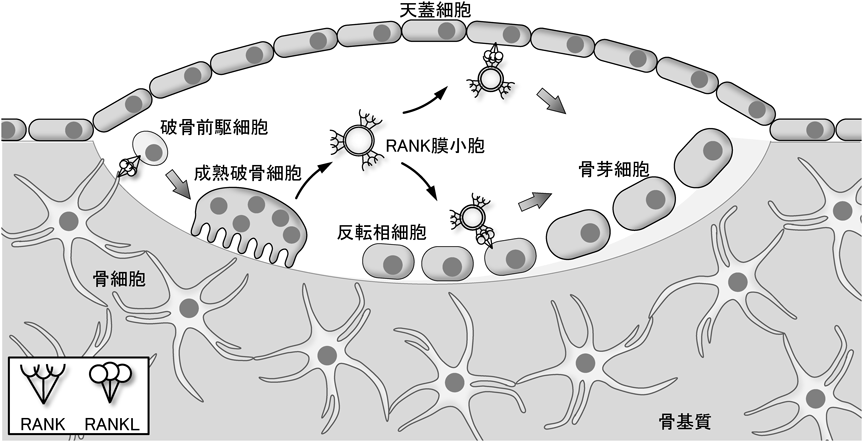
骨リモデリングにおけるRANKLの役割Roles of RANKL in the bone remodeling process
東京大学医学部附属病院・薬剤部Department of Pharmacy, The University of Tokyo Hospital ◇ 〒113–8655 東京都文京区本郷7–3–1 ◇ 7–3–1 Hongo, Bunkyo-ku, Tokyo 113–8655, Japan
発行日:2019年8月25日Published: August 25, 2019