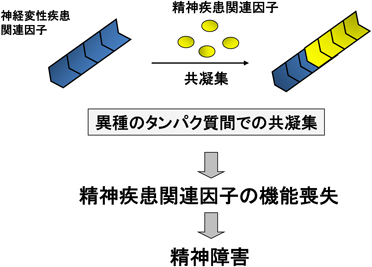
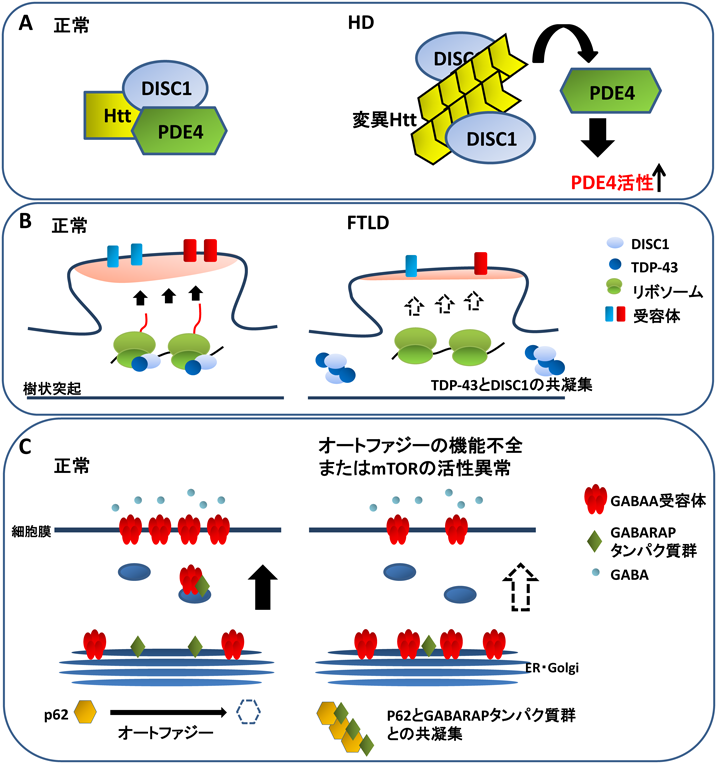
神経変性疾患と精神疾患をつなぐ分子機構の解明—タンパク質の凝集化による精神障害の発現—Molecular mechanisms underlying mental disorders in neurodegenerative diseases
国立研究開発法人理化学研究所,脳神経科学研究センター,タンパク質構造疾患研究チームRIKEN, Center for Brain Science, Laboratory for Protein Conformation Diseases ◇ 〒351–0198 埼玉県和光市広沢2–1 ◇ 2–1 Hirosawa, Wako, Saitama, 351–0198
発行日:2019年8月25日Published: August 25, 2019