パーキンソン病は,黒質のドーパミン作動性ニューロンの喪失によって引き起こされる運動症状を主な特徴とする神経変性疾患である(近年では,運動症状以外にも種々の症状を呈することが知られている).パーキンソン病のほとんどの症例は高齢者に散発的に発生するが[一般的に“パーキンソン病”といわれる孤発性パーキンソン病(孤発性PD)],症例の約5~10%は常染色体上の遺伝子変異によって引き起こされる遺伝性パーキンソン病である[正式には“遺伝性パーキンソニズム”であるが,わかりやすさを優先して本稿では“遺伝性パーキンソン病(遺伝性PD)”と記載する].遺伝性PDには,孤発性PDと比較して極端な若年で病状が観察される若年発症型のPDが含まれる.
PARK21)およびPARK62)遺伝子は若年で発症する遺伝性潜性PDの原因遺伝子である.遺伝様式や,欠失を含むゲノム変異の様式から,PARK2にコードされるユビキチン連結酵素(ユビキチンリガーゼ:E3)Parkinや,PARK6にコードされるプロテインキナーゼPINK1の機能喪失によって病気が発症することが明らかになっている.また膨大な論文から,少なくとも培養細胞においては,ParkinおよびPINK1が脱分極したミトコンドリア(つまり機能障害のあるミトコンドリア)に対するオートファジー(マイトファジー)を誘導して,ミトコンドリアの品質管理を行うことが示されている3).
一方,PINK1とParkinは上述のようにミトコンドリアをマイトファジー分解に導く機能を持つので,通常時にはその機能には何重にも抑制がかかっている.たとえば,PINK1は通常時には分解されて細胞内に存在しないようになっている.Parkinは通常時はサイトゾルに存在し,ミトコンドリア上には存在しない.さらに,Parkinは常に自己阻害プロセスによってE3としての酵素活性を失っている.しかしながらミトコンドリアの膜電位が低下すると,Parkinはサイトゾルからミトコンドリアに移行するとともに,そのE3機能が活性化状態に変換されて,マイトファジーを誘導する.なお,このParkinの移行と活性化のプロセスは,損傷ミトコンドリアの外膜上で分解を免れて蓄積し活性化されたPINK1によって開始される.
PINK1やParkinの機能を考えるためには,両者の機能がオンになる分子メカニズムの詳細な理解が必要不可欠である.また,PINK1やParkinを活性化できれば,ミトコンドリアの代謝回転が増加して遺伝性PDの進行を妨げる可能性が示唆されているが,そのような治療を念頭に置いた研究を進める上でも,両者の活性化メカニズムの理解は重要である.
2. 構造解析から示されたParkin不活性化のメカニズム
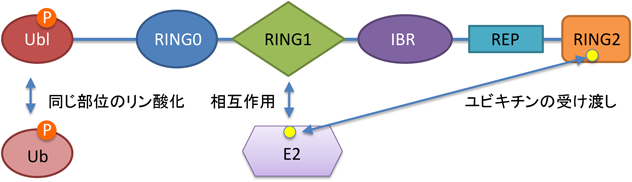
Parkinは,N末端にUblドメイン(Ubiquitin like domain)を持ち,その後にリンカーを介して四つのZn2+結合ドメイン(RING0, RING1, IBR,およびRING2)を持つE3である(図1).IBRとRING2の間には構造解析から明らかにされたRING1に結合する領域があり,REPドメインと呼ばれる(REPリンカーと呼ぶ研究者もいる).
ユビキチンは基本的にE1, E2, E3の働きを経て基質へと付加される.ParkinのRING1はE2結合部位,RING2は活性中心部位である.ParkinがE3として機能する際には,まずParkinのRING1に結合したE2からRING2に存在する酵素活性中心システイン(C431)へとユビキチンがチオエステル結合の状態で転移し,その後にRING2から基質のリシン残基にユビキチンが転移すると考えられている(図1)4).
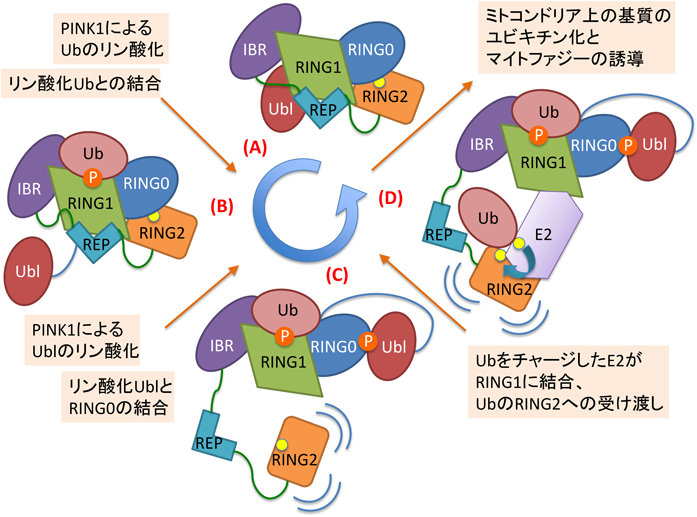
Parkinの最初の結晶構造(Parkin単独の結晶構造)が明らかにされたのは2013年であり,その分子構造からParkinが複数の自己抑制メカニズムを持つことが明瞭に示された5–7).まず,当時解明されたParkinの構造をドメイン間の位置関係から述べると,「RING0ドメインとRING2ドメインが結合し,UblドメインとRING1ドメインが結合している」ことが示された(模式図を図2Aに示す).
ParkinのRING1ドメイン上にはE2結合部位が存在するが,興味深いことに,UblドメインおよびREPドメインが立体的に“RING1ドメイン上のE2結合部位にE2が結合すること”を妨げている.また上述のようにParkinはRING2ドメイン内にある活性中心Cys(C431)を用いて,E2酵素からユビキチンを高エネルギー状態(チオエステル結合)で受け取るが,驚くべきことに,この触媒中心であるRING2ドメインのC431もRING0ドメイン[unique Parkin domain(UPD)ドメインとも呼ばれる]との結合によって外からアクセスできないように塞がれている(図2A).このように,Parkinを不活性化状態に保つメカニズムは,「Parkinに不活性化因子が結合することに由来する」のではなくて,「Parkinの分子内で引き起こされる構造的な自己阻害に由来する」ものであった.
3. Parkin活性化因子としてのリン酸化ユビキチン
Parkinの活性化メカニズムに大きな進展があったのは2012年から2014年である.ParkinのE3酵素活性は完全にPINK1キナーゼに依存している.2012年から2013年にかけて,PINK1がParkinのUblドメインの65番目のセリン(S65)をリン酸化することが示された8–10).ただし,ParkinのS65A変異体のE3活性が完全に失われるわけではなく,またParkinのリン酸化模倣変異(S65E変異など)によってParkinが活性型に変換されるわけではないことから,PINK1の基質はParkinだけではないことが予想された.2014年にPINK1のもう一つの決定的に重要な基質がユビキチンであり,PINK1によって65番目のセリン(S65)がリン酸化された“リン酸化ユビキチン(PhosUB)”がParkinの活性化因子であることが明らかにされた11–13).なお,紙面の都合で詳細は他の総説にゆずるが,PhosUBはParkinの損傷ミトコンドリアへの移行にも必須の役割を担っている3).これらの知見を支持するデータとして,たとえば細胞内のすべてのユビキチンを非リン酸化フォーム(S65A)に置き換えると,ミトコンドリアの膜電位を低下させても,ParkinのE3機能の活性化は起こらなくなり,損傷ミトコンドリアへの移行も阻害される14, 15).同様に,Parkinの“リン酸化ユビキチンとの結合部位”に変異を導入すると,ミトコンドリア損傷依存的なParkinの活性化も損傷ミトコンドリアへの移行も観察されなくなる16).
次に興味が持たれたステップは,「どのように,活性化因子であるPhosUBとParkinが結合しているのか」である.驚くべきことに,PINK1キナーゼの基質がユビキチンであることが報告された翌年には,PhosUBと結合したParkinの構造(∆Ubl-Parkin-PhosUB複合体の構造)が解明された17).
ParkinとPhosUBは主にRING1ドメインで結合しており(ただしE2結合領域の逆側からRING1ドメインに結合している;図2Bを参照),その結合によって,RING1ドメインのHelix[phosphoUb binding helix(pUBH)とも呼ばれる]に顕著な構造変化を引き起こし,その結果としてIBRドメインも大きく移動していた.またPhosUBとRING1の結合様式と,UblとRING1の結合様式を比較することによって,PhosUBによってUblドメインとRING1ドメインの結合が阻害されることも示唆されていた(この研究17)で解明されたParkin-PhosUB複合体の構造にはParkinのUblドメインは含まれていないので,上記知見は直接的な証拠というよりも間接的な証明である).まとめると,2014年にParkinとPhosUBの結合がParkinの活性化に重要であることが示されて,2015年の構造解析でも確かにPhosUBの結合でParkinに構造変化が引き起こされることが明確になった17).
このように構造解析が進展する一方で,依然としてPhosUBに結合したParkinにおける活性化の仕組みには謎が残っていた.つまり,Parkin-PhosUB複合体においては,上述のようにRING1ドメインに存在するヘリックスの大きな構造変化などが観察された一方で,RING1のE2結合部位にはREPドメインが結合しており,RING2にある活性中心Cysは(少し構造が緩んでいると思われるが)依然としてUPDによって容易にアクセスできない抑圧状態であった(図2B).つまり,ParkinとPhosUBの結合はParkinのE3機能の活性化に必須であるが,両者が結合しさえすればParkinの自己阻害が完全に解消されるわけではなく,活性化のためにはさらなるステップが存在する可能性が残っていた.
この謎を解いたのが,Komanderのグループ,あるいはTrempeとGehringのグループによる2018年のリン酸化Parkinの構造解析論文である18, 19).以下にその詳細を解説する.
4. Parkin活性化のメカニズム:Komanderらによる構造解析
Komanderのグループは,2013年の不活性型Parkinの構造解析5)や,2015年のParkin-PhosUB複合体の構造解析17)も行っているが,彼らの構造解析には常に多くの工夫が凝らされており,それは今回の論文18)でも同じである.ParkinのUblドメインとRING0/UPDドメイン間にはフレキシブルなリンカー領域が存在するが,このリンカー領域のアミノ酸配列をみると,高度に保存された部分と,生物種によって非常に異なる部分とを含んでいる.一般に,進化的な保存度が低く(=重要性が低く),移動の自由度の高い領域は結晶形成を阻害する傾向がある.さまざまな種類の動物のParkinの配列を調べると,ガータースネーク(Thamnophis sirtalis)というヘビのParkin(TsParkin)のリンカー配列が最もコンパクトな短いものであった.そのために,Komanderらは活性型Parkinの構造解析の対象としてガータースネーク由来のTsParkinを選択した.
さらに彼らは,活性型Parkinの結晶解析が移動自由度の高いRING2ドメインによって妨げられている可能性に気づいた.それならば「RING2ドメインを除去したParkinの構造解析」を行えばよいと思われるが,TsParkinはRING2ドメインなしで発現されると不溶性であった.RING2とRING0/UPDは主に疎水領域を介して結合しているので,おそらくRING2がないとRING0/UPDの疎水性領域が露出されてしまい,その結果として不溶性になってしまうことが考えられた.そこでKomanderらは,まずTEV(Tobacco etch virus)プロテアーゼ切断部位をTsParkinのIBR-とRING2間のリンカー領域に導入し,TEVプロテアーゼによってRING2が切除できるParkinを可溶性の状態で得た[TsParkin(IBR-TEVsite-RING2)].次にUb-C3Brというユビキチンプローブ(C末端に活性基のついたユビキチン)と上記のTsParkin(IBR-TEVsite-RING2)をGST-PINK1存在下で反応させた.PINK1がUb-C3Brをリン酸化し,リン酸化されたPhosUB-C3Brが活性基(C3Br)を介してParkinと安定な複合体を形成するので,この反応によって「PhosUBが安定して結合したTsParkin(IBR-TEVsite-RING2)」が得られた.また,3節に記載したように(図2Bの模式図も参照),PhosUBの結合によってParkinのUblドメインがRING1ドメインから外れやすくなるので,このRING1ドメインからリリース(放出)したUblドメインが反応液に含まれるGST-PINK1によって効率よくリン酸化される.こうした試料調製時の工夫によって,Ublドメインがリン酸化されたTsParkin(IBR-TEVsite-RING2)を得て,その後にTEVプロテアーゼを用いて自由度の高い(=それゆえに結晶化を邪魔していると思われた)RING2ドメインを除去することが可能になった.今回,構造解析されたのは,このような手順を経て準備された「PhosUBと結合し,Ublドメインがリン酸化されたTsParkin∆RING2」である.なお水素–重水素交換(HDX)-MSプロファイルの比較(詳細は6節に記載)などから,Komanderらは「RING2を除去しても,その操作が残った部分に影響を与えて,TsParkin本来の構造を失わせている可能性はない」と述べている.
得られたUblがリン酸化されたTsParkin∆RING2とPhosUBの複合体(以降はPhosParkin∆2-PhosUBと記載)の結晶構造は,1.8 Å解像度で以下の知見をもたらした.
まず,2013年に報告されている全長Parkinの構造(X)や,2015年に報告されているParkin-PhosUB複合体(非リン酸化Parkinとリン酸化ユビキチンの複合体,ただし実際に決められた構造にはUblドメインが含まれていない)の構造(Y)と,今回新しく決定されたPhosParkin∆2-PhosUB(リン酸化Parkinとリン酸化ユビキチンの複合体,ただしRING2ドメインはない)の構造(Z)を比較すると,Parkinのコアとなる部分の構造(RING0/UPD-RING1-IBR)は(Y)–(Z)間でほとんど同じであり,またParkinを構成するモジュールである個々のドメインの構造も,(X)–(Y)–(Z)間でほとんど同じであった.では,何が大きく違うかというと,個々のドメインの位置関係,具体的にはUblドメインの位置がまったく異なっていたのである.
2013年に報告されていた不活性型Parkinの構造(X)では,図2Aに模式的に示すようにUblドメインはRING1ドメインと結合していたが,今回構造解析されたPhosParkin∆2-PhosUBの構造(Z)では,図2Cに模式的に示すように,リン酸化Ublドメイン(PhosUbl)はRING0/UPDドメインに結合していた.両者のUblドメインの位置を比較すると,実に50 Å以上も移動している.PhosUblとRING0/UPDとの間の相互作用部位をみると,Ublドメインのリン酸化Ser65は,RING0/UPD上のK161, R163, K211によって形成される正電荷を帯びたポケットに収まっている.実はK161とK211はParkinの機能に重要な残基としてよく知られていた.というのも,K161とK211は遺伝性PD患者にみられる二つの有名な突然変異(K211NおよびK161N)部位であり,培養細胞を用いたさまざまなアッセイでK211NとK161NがParkinの機能を失わせることが報告されていたからである.実際,K211N変異を有するリン酸化Parkinは,ユビキチン由来の活性化プローブであるUb-VSによって修飾されず(=活性型に変換されないことを示唆する),またHDX-MS解析(後述)でもRING2が放出された兆候をほとんど示さなかった.これらの結果は,PhosUblが,RING0/UPDドメイン中のK211が形成する正電荷を帯びたポケットと相互作用できないと,ユビキチンを受け取るRING2ドメインの活性中心Cysがアクセス不能のままであることを示している.
さらに今回のKomanderらの構造解析からはRING0/UPDドメインと結合する新たな領域が発見され,RING2とRING0/UPDの結合を阻害する新たな仕組みの存在が示唆された.先述のように,RING0/UPDドメインとPhosUblドメインが結合すると,立体障害が生じるために,自動的にRING2ドメインはRING0/UPDドメインと結合できなくなることが予想される.このことに加えて,TsParkinにおいてはUblドメインの後ろに続くリンカー領域中のL102–L107の部分が,RING0/UPDドメインとRING2ドメインの結合ポケットに相当する部分に入り込んでいた.そこで,Komanderらは新たにこのL102–L107近傍の部分をactivation element(ACT element)と命名し,このACT elementがRING0/UPDドメインとRING2ドメインの結合を阻害していることを提唱した.実際に,PD患者でもこのACT element中に変異が見いだされており(R104W),またリン酸化ParkinにR104A変異を導入するとユビキチン由来の活性化プローブであるUb-VSによって修飾されない(=活性型に変換されない)ことが示されている.
ただし,このACT elementは,アミノ酸配列[SLTR-疎水性(V/I/L)-DLSXX]でみるとヒト・マウス・ヘビ・ゼブラフィッシュなどで保存されているが,次節で紹介するミカンコミバエのParkin(BdParkin)では保存されていない.それにもかかわらず,ACT elementのないミカンコミバエのBdParkinも明瞭な“Ublドメインのリン酸化に依存したE3の活性化”を示すのは不思議である.ACT elementがParkin活性化に際して必須の役割を担っているのかについては,今後の展開を注視する必要がある.
5. Parkin活性化のメカニズム:TrempeとGehringによる構造解析
一方のTrempeとGehringのグループも,2013年に不活性型Parkinの構造を報告する6)など,Parkinの構造解析で成果をあげてきた研究者である.今回,彼らはPhosUBとE2酵素(UbcH7)との複合体として,リン酸化された活性型Parkinの構造を報告している19).
活性化Parkinのタンパク質結晶を得るために,彼らはさまざまなParkinのコンストラクトをスクリーニングした.最も解析に優れた結晶は,ヒトParkinとアミノ酸配列で43%の同一性を示すミカンコミバエ(Bactrocera dorsalis)のParkin(BdParkin)で得られた.さらに彼らはE2酵素との共結晶化を促進するために,ヒトのE2であるUbcH7とBdParkinをつなげた融合タンパク質を作製した(強引なやり方に思えるが,このようなアプローチは他のE2-RING複合体の結晶化において使用されてきた手法である).BdParkinには,Ublの前にN末端延長部分があるが,この部分は削除された.彼らは長さの異なるリンカーを介してUbcH7とBdParkinを融合し,PhosUBの存在下で結晶化のさまざまな条件を検討した.こうして結晶化の実験を進める過程で,Komanderらの場合と同様に,TrempeとGehringも融合タンパク質からREPリンカーとRING2ドメインを削除した方がよいことに気づいた.前節で記載したように,不活性型のParkinはRING2ドメインなしでは溶解性が乏しくなるが,TrempeとGehringらはREP-RING2を欠いたBdParkinタンパク質をPINK1およびユビキチンと同時発現させることで,可溶性を維持できたようである.このようにして,彼らはリン酸化ユビキチン(PhosUB)・E2(UbcH7)・リン酸化BdParkinの三者複合体の構造を明らかにした.
TrempeとGehringのBdParkinの構造解析からも,Parkinの活性化はUblドメインの位置を大きくシフトさせることが示されている.自己阻害された不活性型のParkinにおいて,UblドメインはRING1ドメインに結合しているが(図2A),彼らの構造においても,リン酸化されたUblドメインはRING0ドメインに移動(結合)している.この立体配座では,RING1上のE2結合部位が利用可能になる一方で,リン酸化Ubl(PhosUbl)と結合部位を競合するRING2はRING0に結合することができない(図2Dに模式的に示す).また,Ubl中のリン酸化S65(pS65)は,正に帯電した二つのリシン(K161, K211)と一つのアルギニン(R163)によって安定化されている.このあたりの結論は,文献18, 19でまったく一緒である.
BdParkinの結晶構造で観察された「PhosUblとRING0の結合」がミカンコミバエ由来Parkin(BdParkin)特異的な現象である可能性や,E2とParkinを融合したタンパク質を使用したことによるアーティファクトである可能性を除外するために,TrempeとGehringのグループはラットParkinを用いた検証実験を行っている.K161, R163,およびK211を個別にアスパラギンに変異させ,変異タンパク質をPINK1によってリン酸化した.これらのリン酸化–変異Parkinタンパク質を精製して自己ユビキチン化アッセイにかけると,三つの変異体は劇的なE3活性の減少を示した.つまり,K161N変異体はいくらかの残存活性を示したが,R163NおよびK211N変異体は完全に不活性であった.これらの変異の影響が「Parkinがリン酸化を受けなくなった」ことに由来する可能性を排除するために,Phos-tag SDS-PAGEや質量分析を行った結果,三つの変異体はすべて完全にリン酸化されていることが確認された.
もっとも,4節で述べたようにK161NやK211Nは有名な遺伝性PD患者由来の変異であり,すでにParkinのE3活性低下も含めたさまざまな異常が報告されていた.したがって,「K161NやK211NでE3機能が阻害される」こと自体に新規性があるわけではなく,「K161NやK211NのE3機能阻害が,PhosUblとRING0が結合できないことに由来すると解釈できる」ところに新規性がある.
Komanderらの研究(4節)では行われていない,TrempeとGehringによるユニークな解析として,彼らはNMR実験によってもRING0がPhosUblに結合することを確認している.15NでラベルしたPhosUblはNMR中で完全にアサインされた15N-1Hスペクトルを示すが,このときに溶液中に∆Ubl-Parkinを加えると,Ublドメインのリン酸化セリン(pS65)に由来するシグナルが完全に消失する.対照的に,溶液中に∆Ubl-Parkin(K211N)を加えても,PhosUblドメインのpS65に由来するNMRのシグナルは消失しない.このことは,溶液中でPhosUblドメインのpS65が,RING0ドメイン中のK211依存的に∆Ubl-Parkinと結合しており,その結果としてシグナルが消失することを示している.
さらに溶液中の分子間相互作用(上述のようにUblと∆Ubl-Parkinを混ぜ合わせて,相互作用をみるような実験系)ではなくて,同一の分子内での相互作用を調べるために,彼らはsortase A protein-ligation systemを用いて「Ublドメインのみが15NでラベルされたParkin」を作製し,Ublドメイン部分のNMRスペクトラムを解析する実験も行っている.その結果,1)Parkin(全長)に含まれる15N-ラベルPhosUblのシグナルは,15N-ラベルPhosUblドメイン単独のシグナルより顕著に減衰しているが,2)K211N変異体中の15N-ラベルPhosUblのシグナルは,遊離の15N-ラベルPhosUblドメインのシグナルとほぼ同じであることを見いだした.これらの結果は,「一つのParkin分子内でも,pUblドメインがK211依存的にRING0と結合するためにシグナルが減弱する」と考えると矛盾なく説明可能なデータである.
まとめると,二つの構造解析の論文18, 19)からみえてくるのは,Parkinの活性化に際して,UblドメインがRING1と結合してParkinのリン酸化と活性化を抑える“阻害的な位置”(図2A)から,リン酸化されたのちにRING0/UPDと結合してRING2を放出させる“活性化を促す位置”(図2C, D)へと劇的に再配置されていることである.
6. 溶液中の水素–重水素交換(HDX)-MSによるParkin活性化に伴うドメイン再配置の検討
このように結晶構造解析から「活性型Parkinが大規模な構造変化とドメインの構成変化を起こしている」ことが示されたが,それは静的な結晶中で「活性化した後の構造」データであり,Parkinが溶液中で時間依存的にそのような構造変化を起こしていることを示すデータではない.Komanderのグループ18),TrempeとGehringのグループ19),双方が水素–重水素交換(HDX)-MSを用いてこの問題点の解決に挑んでいるので,最後にその結果を紹介したい.
HDX-MSはタンパク質主鎖のアミド水素「[R–(C=O)–NH–R′]のNH部分の水素」と溶液中の重水素との相対交換速度を調べる技法である.タンパク質の中には「時間が経過しても溶媒中の重水素との交換がほとんどない」ペプチド(領域)と,「時間の経過とともに溶媒中の重水素との交換が起こる,または交換が増加する」ペプチド(領域)とが存在するが,交換の遅い前者は「相対的にタンパク質のコアの中にある(したがって溶媒とのアクセスが困難な)領域」であり,交換が進む後者は「相対的にタンパク質の表面に露出した領域」である.HDX-MSは主鎖アミド水素と溶液中の重水素との相対交換速度を調べることで,フォールディングしたタンパク質における溶媒への接近しやすさを評価することができる.この方法の威力は,活性化の過程を通じて「同一サンプル内のペプチドと溶媒との関係性」を比較できる能力にあり,タンパク質の中で溶液中に新たに露出される領域や,別なドメインと新たに結合して保護される領域を見積もることができる.
HDX-MS実験では,解析時のペプチドの被覆率(coverage)に依存する分解能で重水素交換の速さをマッピングすることが可能であり,TrempeとGehringの論文ではその分解能が15アミノ酸に相当する.TrempeとGehringらはラットのParkinをHDX-MS解析し,KomanderらはヒトParkinをHDX-MS解析しているが,両者がPhosUBとの結合・Ublドメインのリン酸化・PhosUbl結合部位への変異の導入,などの影響を調べた結果は以下のとおりである.
まず自己阻害された不活性型のParkinをHDX-MS分析すると,Ublドメインとのリンカー領域の一部の重水素化のレベルが他の領域に比較してやや高いが,全体として重水素化は低く抑えられている.そこにPhosUBを添付すると,Ublドメインの重水素化が促進される.この結果は,2015年の結晶構造解析や,in vitroのリン酸化実験からも示唆されていたことであり,「PhosUBが結合すると,ParkinのUblはRING1と結合できなくなる(その結果,溶媒との接触が増える)」という予想と一致する(図2Aから図2B).PhosUB添付の効果はIBRドメインにおいても観察されており,残基323~338を含む領域の重水素化は不活性型Parkinと比較してむしろ減弱する.一方で,PhosUBの添付では,活性中心であるRING2の重水素化にはほとんど変化が観察されなかった.
HDX-MS解析で最も大きな変化が観察されたのは,Parkinをリン酸化した後のRING2ドメインである.つまり,Parkinのリン酸化によって,RING2のC末端へリックスの重水素化が劇的に増加することが観察された.2013年に報告された不活性型のParkinの構造では,RING2のC末端ヘリックスはRING0にしっかりと結合しており(図2A),溶媒から保護されているので,重水素化は阻害されているはずである.一方で,Parkinがリン酸化されると,PhosUblがRING0に結合することで,RING2は放出されるが,その結果としてRING2のヘリックスは完全に溶媒中に露出されて,急速に重水素と交換されると期待される(図2C).したがって,このHDX-MSの結果は,「Ublのリン酸化に依存したRING2の放出によってParkinが活性化される」という結晶構造から導かれたモデルとよく一致していることになる.
さらに確認のために,TrempeとGehringらは先述のK211N変異体を用いたHDX-MS解析も行っている.非リン酸化状態では,野生型ParkinとK211N変異体の重水素交換速度は区別できない状態(重水素交換のパターンは同じ状態)であった.しかしながら,リン酸化後の効果は劇的に異なっていた.つまり野生型Parkinではリン酸化後にRING2の重水素交換速度が増加するが,K211N変異体ではリン酸化後のRING2の重水素交換速度の増加が完全に阻害された.
一方,Ublドメインの重水素交換速度は,二つのグループで実験手法が異なる部分もあって単純に比較するのは少し難しい.TrempeとGehringの論文では,短いタイムポイントではリン酸化Parkin(K211N)のUbl(42~61)ペプチドの重水素交換速度は野生型よりも高いと記載されている[これはPhosUblがRING0(K211N)に結合できないために,溶液中に存在する時間が長いためと理解できる].Komanderの論文では,Parkinのリン酸化だけではUblドメインの重水素交換はあまり変化せず,E2とUbの複合体(non-dischargeable E2-ubiquitin結合体)を試料に添加することで,PhosUblの重水素交換速度が顕著に減少すると報告している(これは図2Dで示すように,最終形態としてPhosUblがRING0と安定して結合すると溶媒との交換が起こらなくなるため,と理解できる).
いずれにせよHDX-MS解析からは,ParkinのUblドメインはK211N変異体を用いると自由度が増加し,RING2ドメインはParkinのリン酸化・活性化に伴って自由度が増すという,結晶構造と一致する知見が得られている.
本稿では,今まで日本語総説等で詳しく紹介されたことがないと思われる,構造解析を基盤とするParkin活性化の分子メカニズムを解説した.もっとも,本稿では2報の論文18, 19)の著者らの主張に従って,あたかもParkin活性化メカニズムのすべてが解明されたかのように記述をしたが,実際には不明な点も残されている.たとえばParkin S65A変異体の表現型は解釈が難しい.仮に“PhosUblがRING0に結合してRING2を弾き飛ばすことがParkin活性化の本質”なのであれば,Ublドメインのリン酸化が停止するS65A変異によってParkinの機能は完全に失われるはずである.しかしながら,実際にはParkin S65A変異体でもcell free assayではE3活性が残っているし,ParkinのE3活性に依存したミトコンドリアへの移行も観察される(つまり,S65A変異体は,細胞内でそれほど劇的な表現型を示すわけではない)13).もっといえば,Parkin ∆Ublドメインでも細胞内のParkinの機能が完全に失われるわけではない13).このあたりの矛盾点が今後どのように解決されるのかは興味深い.ただし,上述のような謎は残っているが,本稿で紹介した2報の論文18, 19)がParkin活性化に関して多くの有益な情報を提供してくれたのは紛れもない事実である.
最後にもう一つ言及したいことは,今回の構造解析の結果と,Parkinのユニークな「基質選択性の低さ・触媒するユビキチン鎖の多様性の高さ」とは相性がよいことである.
ユビキチン鎖にはいくつかの種類があるが,Parkinによって損傷ミトコンドリアに付加されるのはK6鎖,K11鎖,K48鎖,K63鎖であることが報告されている20–23).Parkinが損傷ミトコンドリアにK63鎖を付加することは,(1)質量分析装置を用いた実験,(2)特定の種類のユビキチン鎖を検出できるプローブを用いた実験,(3)ユビキチン鎖の種類に特異的な抗体を用いた実験,など複数の手法によって確認されており,信頼度が高い.一方で,Parkinが損傷ミトコンドリアにK48鎖を付加することも,(1)質量分析装置を用いた実験,(2)プロテアソーム阻害剤を用いた実験,などから確認されており,Parkinが複数のタイプのユビキチン鎖を触媒する可能性が示されている(ただし,マイトファジーの誘導という役割においては,K63鎖の貢献が大きいと思われる).ここで一度Parkinの構造に立ち返ると,活性中心を含むRING2触媒ドメインは,RING0から解放された後は約30アミノ酸のIBR-RING2リンカーによってのみ他のドメインに結合されており(図2C),可動性が非常に高いと予想される.一方でK63ユビキチン鎖や直鎖ユビキチン鎖が選択的に形成されるときには,ドナーユビキチンとアクセプターユビキチンが正しく配置される必要がある24–26).そのことを考えると,Parkinの活性中心(RING2)の自由度の高さは「特定の種類のユビキチン鎖」を形成するにはいかにも不適当であり,逆に活性中心の自由度の高さゆえに多様なユビキチン鎖を形成するのに適しているように思われる.
同様に,Parkinの基質選択性の低さは以前から報告されているが27),その背景は不明であった.しかしながら,「ユビキチンと高エネルギーなチオエステル結合を形成した活性中心(RING2)部分がフレキシブルな可動領域として存在する」活性型Parkinの構造(図2D)をイメージすると,“基質との相互作用”よりも“近傍に存在すること”が基質選択性を決定する27)という仮説の信憑性が増してくる.実際に研究をしていると,Parkinは損傷ミトコンドリア上の外膜タンパク質であれば何でもユビキチン化する印象を受ける.このようなE3としては例外的な基質特異性の低さも,活性化されたParkinの構造をみると,妥当に思えてくるものである.
謝辞Acknowledgments
原稿の作成に際して,原図の作成と多くの有益なコメントをしてくれた東京大学農学部昆虫遺伝研究室の松田(今井)典子博士に深謝いたします.
引用文献References
1) Kitada, T., Asakawa, S., Hattori, N., Matsumine, H., Yamamura, Y., Minoshima, S., Yokochi, M., Mizuno, Y., & Shimizu, N. (1998) Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature, 392, 605–608.
2) Valente, E.M., Abou-Sleiman, P.M., Caputo, V., Muqit, M.M., Harvey, K., Gispert, S., Ali, Z., Del Turco, D., Bentivoglio, A.R., Healy, D.G., et al. (2004) Hereditary early-onset Parkinson’s disease caused by mutations in PINK1. Science, 304, 1158–1160.
3) Yamano, K., Matsuda, N., & Tanaka, K. (2016) The ubiquitin signal and autophagy: an orchestrated dance leading to mitochondrial degradation. EMBO Rep., 17, 300–316.
4) Wenzel, D.M., Lissounov, A., Brzovic, P.S., & Klevit, R.E. (2011) UBCH7 reactivity profile reveals parkin and HHARI to be RING/HECT hybrids. Nature, 474, 105–108.
5) Wauer, T. & Komander, D. (2013) Structure of the human Parkin ligase domain in an autoinhibited state. EMBO J., 32, 2099–2112.
6) Trempe, J.F., Sauve, V., Grenier, K., Seirafi, M., Tang, M.Y., Menade, M., Al-Abdul-Wahid, S., Krett, J., Wong, K., Kozlov, G., et al. (2013) Structure of parkin reveals mechanisms for ubiquitin ligase activation. Science, 340, 1451–1455.
7) Riley, B.E., Lougheed, J.C., Callaway, K., Velasquez, M., Brecht, E., Nguyen, L., Shaler, T., Walker, D., Yang, Y., Regnstrom, K., et al. (2013) Structure and function of Parkin E3 ubiquitin ligase reveals aspects of RING and HECT ligases. Nat. Commun., 4, 1982.
8) Shiba-Fukushima, K., Imai, Y., Yoshida, S., Ishihama, Y., Kanao, T., Sato, S., & Hattori, N. (2012) PINK1-mediated phosphorylation of the Parkin ubiquitin-like domain primes mitochondrial translocation of Parkin and regulates mitophagy. Sci. Rep., 2, 1002.
9) Kondapalli, C., Kazlauskaite, A., Zhang, N., Woodroof, H.I., Campbell, D.G., Gourlay, R., Burchell, L., Walden, H., Macartney, T.J., Deak, M., et al. (2012) PINK1 is activated by mitochondrial membrane potential depolarization and stimulates Parkin E3 ligase activity by phosphorylating Serine 65. Open Biol., 2, 120080.
10) Iguchi, M., Kujuro, Y., Okatsu, K., Koyano, F., Kosako, H., Kimura, M., Suzuki, N., Uchiyama, S., Tanaka, K., & Matsuda, N. (2013) Parkin-catalyzed ubiquitin-ester transfer is triggered by PINK1-dependent phosphorylation. J. Biol. Chem., 288, 22019–22032.
11) Koyano, F., Okatsu, K., Kosako, H., Tamura, Y., Go, E., Kimura, M., Kimura, Y., Tsuchiya, H., Yoshihara, H., Hirokawa, T., et al. (2014) Ubiquitin is phosphorylated by PINK1 to activate parkin. Nature, 510, 162–166.
12) Kazlauskaite, A., Kondapalli, C., Gourlay, R., Campbell, D.G., Ritorto, M.S., Hofmann, K., Alessi, D.R., Knebel, A., Trost, M., & Muqit, M.M. (2014) Parkin is activated by PINK1-dependent phosphorylation of ubiquitin at Ser65. Biochem. J., 460, 127–139.
13) Kane, L.A., Lazarou, M., Fogel, A.I., Li, Y., Yamano, K., Sarraf, S.A., Banerjee, S., & Youle, R.J. (2014) PINK1 phosphorylates ubiquitin to activate Parkin E3 ubiquitin ligase activity. J. Cell Biol., 205, 143–153.
14) Okatsu, K., Koyano, F., Kimura, M., Kosako, H., Saeki, Y., Tanaka, K., & Matsuda, N. (2015) Phosphorylated ubiquitin chain is the genuine Parkin receptor. J. Cell Biol., 209, 111–128.
15) Ordureau, A., Heo, J.M., Duda, D.M., Paulo, J.A., Olszewski, J.L., Yanishevski, D., Rinehart, J., Schulman, B.A., & Harper, J.W. (2015) Defining roles of PARKIN and ubiquitin phosphorylation by PINK1 in mitochondrial quality control using a ubiquitin replacement strategy. Proc. Natl. Acad. Sci. USA, 112, 6637–6642.
16) Yamano, K., Queliconi, B.B., Koyano, F., Saeki, Y., Hirokawa, T., Tanaka, K., & Matsuda, N. (2015) Site-specific Interaction Mapping of Phosphorylated Ubiquitin to Uncover Parkin Activation. J. Biol. Chem., 290, 25199–25211.
17) Wauer, T., Simicek, M., Schubert, A., & Komander, D. (2015) Mechanism of phospho-ubiquitin-induced PARKIN activation. Nature, 524, 370–374.
18) Gladkova, C., Maslen, S.L., Skehel, J.M., & Komander, D. (2018) Mechanism of parkin activation by PINK1. Nature, 559, 410–414.
19) Sauve, V., Sung, G., Soya, N., Kozlov, G., Blaimschein, N., Miotto, L.S., Trempe, J.F., Lukacs, G.L., & Gehring, K. (2018) Mechanism of parkin activation by phosphorylation. Nat. Struct. Mol. Biol., 25, 623–630.
20) Ordureau, A., Sarraf, S.A., Duda, D.M., Heo, J.M., Jedrychowski, M.P., Sviderskiy, V.O., Olszewski, J.L., Koerber, J.T., Xie, T., Beausoleil, S.A., et al. (2014) Quantitative proteomics reveal a feedforward mechanism for mitochondrial PARKIN translocation and ubiquitin chain synthesis. Mol. Cell, 56, 360–375.
21) van Wijk, S.J., Fiskin, E., Putyrski, M., Pampaloni, F., Hou, J., Wild, P., Kensche, T., Grecco, H.E., Bastiaens, P., & Dikic, I. (2012) Fluorescence-based sensors to monitor localization and functions of linear and K63-linked ubiquitin chains in cells. Mol. Cell, 47, 797–809.
22) Michel, M.A., Swatek, K.N., Hospenthal, M.K., & Komander, D. (2017) Ubiquitin linkage-specific affimers reveal insights into K6-linked ubiquitin signaling. Mol. Cell, 68, 233–246.e5.
23) Okatsu, K., Saisho, K., Shimanuki, M., Nakada, K., Shitara, H., Sou, Y.S., Kimura, M., Sato, S., Hattori, N., Komatsu, M., et al. (2010) p62/SQSTM1 cooperates with Parkin for perinuclear clustering of depolarized mitochondria. Genes Cells, 15, 887–900.
24) Lechtenberg, B.C., Rajput, A., Sanishvili, R., Dobaczewska, M.K., Ware, C.F., Mace, P.D., & Riedl, S.J. (2016) Structure of a HOIP/E2~ubiquitin complex reveals RBR E3 ligase mechanism and regulation. Nature, 529, 546–550.
25) VanDemark, A.P., Hofmann, R.M., Tsui, C., Pickart, C.M., & Wolberger, C. (2001) Molecular insights into polyubiquitin chain assembly: crystal structure of the Mms2/Ubc13 heterodimer. Cell, 105, 711–720.
26) Moraes, T.F., Edwards, R.A., McKenna, S., Pastushok, L., Xiao, W., Glover, J.N., & Ellison, M.J. (2001) Crystal structure of the human ubiquitin conjugating enzyme complex, hMms2-hUbc13. Nat. Struct. Biol., 8, 669–673.
27) Koyano, F., Yamano, K., Kosako, H., Tanaka, K., & Matsuda, N. (2019) Parkin recruitment to impaired mitochondria for nonselective ubiquitylation is facilitated by MITOL. J. Biol. Chem., 294, 10300–10314.
著者紹介Author Profile
 松田 憲之(まつだ のりゆき)
松田 憲之(まつだ のりゆき)東京都医学総合研究所プロジェクトリーダー.理学博士.
略歴2001年東京大学大学院理学系研究科生物科学修了,博士(理学)取得.その後,理化学研究所基礎科学特別研究員,東京都臨床医学総合研究所外部支援研究員,日本学術振興会特別研究員PD, 理化学研究所上級研究員などを経て,08年東京都臨床医学総合研究所研究員,13年より東京都医学総合研究所副参事研究員,14年より同プロジェクトリーダー,現在に至る.
研究テーマと抱負博士号取得後の約20年を振り返ると,研究場所や研究対象が転々としつつも「ユビキチンに関連する研究」という軸を保って研究者人生を送ってきました.今後もユビキチンを軸足に据えてParkinやPINK1の機能解析を行いつつ,パーキンソン病の発症機構解明に貢献できるような新しい研究も始めなければいけないなぁと思っています.
ウェブサイトhttp://www.igakuken.or.jp/project/to-tomin/to-pro20.html