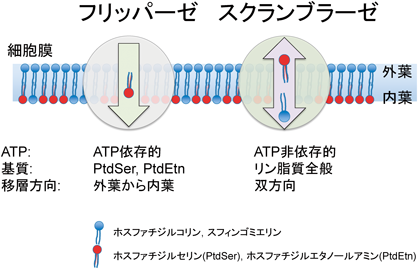
哺乳類細胞の細胞膜は,非対称的なリン脂質二重層で構成される.すなわち,アミノリン脂質であるホスファチジルセリン(PtdSer)やホスファチジルエタノールアミン(PtdEtn)は細胞質に面する内葉に限局し,ホスファチジルコリン(PtdCho)やスフィンゴミエリン(SM)は主に外葉に存在する1).一方,多細胞生物を構成する細胞はさまざまな局面で,この非対称性を崩壊させる2).アポトーシス細胞は,PtdSerを“eat me”シグナルとして細胞表面へ露出し,マクロファージに認識・貪食される3, 4).造血過程で生じる赤芽球から脱核した核や,老化した赤血球などもPtdSer依存的にマクロファージに貪食されると考えられている5, 6).また,出血時に活性化した血小板は,カルシウム依存的に細胞表面へPtdSerを露出する.このPtdSerが血液凝固因子の“足場”として機能することで,血液凝固反応を促進する.これらの反応やシグナル伝達は,細胞膜の内葉に限局するPtdSerが適切な場面で速やかに外葉に移層され,細胞表面に露出されることが鍵となる.しかし,リン脂質の親水基が脂質二重層の疎水領域を横断することはエネルギー的に不利であり,自発的にはほとんど起こらない.したがって,細胞はエネルギー的な障害を回避してPtdSerを移層させる必要がある.PtdSerを移層させる分子として,少なくとも二つの膜タンパク質,フリッパーゼとスクランブラーゼが提唱されている7).フリッパーゼは,PtdSerやPtdEtnを特異的かつATP依存的に,脂質二重層の外葉から内葉へ一方向にフリップする.対照的に,スクランブラーゼはATPを消費することなくリン脂質を非特異的に,脂質二重層の間を双方向にスクランブルする(図1).これらの分子の活性が適切に制御されることにより細胞膜のPtdSerの分布が制御されると考えられていたが,その分子実体や制御メカニズムの多くが不明であった.
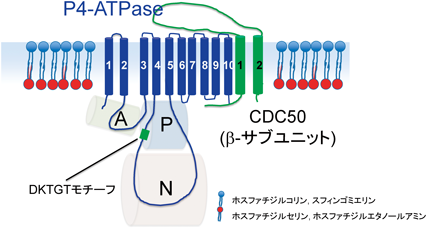
P型ATPaseファミリーに属するP4型ATPase(P4-ATPase)がフリッパーゼとして機能する可能性が提唱されていた.P4-ATPaseファミリーは,真核生物に存在し,進化の過程でメンバーの数が増加している(酵母5,線虫6,マウス15,ヒト14種類)8).P4-ATPaseは他のP型ATPaseと同様に10個の膜貫領域を持ち,分子の中央部分に二つの大きな細胞質ループが存在する.この二つの細胞質ループが,作動領域(A),ヌクレオチド結合領域(N),リン酸化領域(P)を構成する(図2).P型ATPaseの動作原理は,P2-ATPaseファミリーの筋小胞体カルシウムポンプを中心に多くの知見が得られている9).現在,ATPのγ-リン酸基を,P領域に存在するDKTGTモチーフのアスパラギン酸残基に転移させる自己リン酸化と脱リン酸化を繰り返すことにより,構造を変化させながら基質を輸送するという仕組みが考えられている.また,いくつかのP型ATPaseは,サブユニットである他のタンパク質と複合体を形成する.P4-ATPaseも同様に,正しく折りたたまれ,細胞内の適切な場所に輸送されるためには,β-サブユニットとして機能する2回膜貫通タンパク質CDC50との複合体形成が重要であることが報告されている10, 11).これまでP4-ATPaseやCDC50のフリッパーゼとしての機能や細胞における役割は,主に酵母で解析されてきた.当初,酵母のP4-ATPaseであるDrs2pが細胞膜のPtdSerを移層するフリッパーゼであると報告されたが,現在ではDrs2pは主にゴルジ体に局在し,細胞内で機能すると考えられている12).一方で,P4-ATPaseであるDnf1pとDnf2pは酵母の細胞膜に局在するが,主にPtdChoとPtdEtnを移層すると考えられている13).これらの結果と一致するように,酵母の細胞膜ではPtdChoやPtdEtnを移層する活性が強い.一方,哺乳類においても同様の傾向がみられるのか,あるいは細胞膜でPtdSerがフリップされるのか,されるのであればP4-ATPaseのメンバーによるものであるのか,P4-ATPaseが関与する場合どのメンバーが機能するのかなど,その詳細は不明であった.
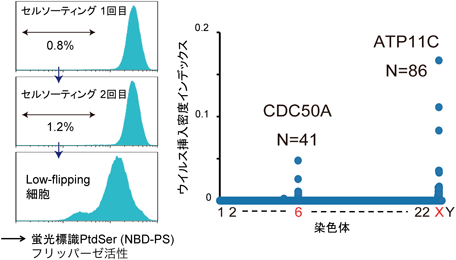
細胞膜におけるフリッパーゼ活性は,蛍光標識したリン脂質プローブ(NBD-lipid)の取り込みで評価することができる.具体的には,エンドサイトーシスなどの非特異的な取り込みを抑えた低温下(15~20°C)で,培養細胞に蛍光標識したプローブを短時間取り込ませ,外葉に残された蛍光プローブをアルブミンなどで除去したのちに,フリップされたプローブの蛍光を測定する.ヒトやマウスの各種血球系細胞株を用いて,細胞膜におけるPtdSer, PtdEtn, PtdCho, SMへのフリッパーゼ活性を評価した結果,例外なくPtdSerを基質として用いた場合に最も強いフリッパーゼ活性が検出された.この結果は,哺乳類細胞の細胞膜にPtdSer-フリッパーゼが存在することを示唆した.そこで,細胞膜で機能するフリッパーゼを網羅的に探索した14).スクリーニングには,Brummelkamp博士が開発した,ヒト一倍体細胞KBM7を用いた遺伝子トラップスクリーニング法を用いた15).現在ではCRISPR-Cas9法を用いたゲノムワイドスクリーニングが可能であるが,当時,上述の方法が哺乳類細胞においてノンバイアスに遺伝子を欠損させる唯一の系であった.KBM7細胞は慢性骨髄性白血病に由来するミエロイド系の細胞株であり,第8染色体を除くすべての染色体が一倍体である16).この一倍体細胞に,強いスプライシングアクセプター配列を組み込んだレトロウイルスを感染させることで,ランダムに遺伝子を欠損させたノックアウト細胞を調製することができる.そこで,KBM7細胞に遺伝子トラップ用レトロウイルスを感染させ,約1億種類の変異細胞を作製した.この変異細胞に蛍光標識したPtdSer(NBD-PS)を添加し,細胞膜におけるPtdSerの取り込みが減少した細胞をセルソーターで回収した.この操作を繰り返すことで,フリッパーゼ活性の減少した細胞(Low-flipping細胞)を濃縮した(図3).Low-flipping細胞のウイルスの挿入位置を,次世代シークエンサーを用いて解析した結果,ヒトP4-ATPaseファミリーに属するATP11Cと,酵母のCDC50のオルソログであるヒトCDC50Aの遺伝子座にレトロウイルスが高頻度に挿入されていることが明らかとなった.実際,ATP11Cを欠損したKBM7細胞はPtdSerを取り込むフリッパーゼ活性が大きく減少し,親株の20%程度にまで減弱した.しかし,依然としてPtdSerは細胞膜の内葉に限局し,非対称的な分布が保持されていた.一方,CDC50Aを欠損したKBM7細胞ではフリッパーゼ活性がほぼ完全に消失し,PtdSerの非対称性を保持することができず,通常の培養条件下においてもPtdSerを露出していた.ヒトやマウスでは10数種類のP4-ATPaseが存在し(Class 1a:ATP8A1, 8A2, Class 1b:ATP8B1~8B4, Class 2:ATP9A, 9B, Class 5:ATP10A~10D, Class 6:ATP11A~11C),ATP9AとATP9Bを除いた大部分のメンバーはCDC50Aと結合することにより小胞体から輸送されることが報告されていた17).実際,EGFPを付加したATP11CはCDC50Aを発現する野生型の血球系細胞では細胞膜に局在するが,CDC50A欠損細胞では小胞体から出ることができなかった.つまり,CDC50AはATP11Cが細胞膜で機能するために必須のサブユニットであり,ATP11Cを含むいくつかのファミリーメンバーを正しく折りたたみ,細胞膜に輸送することで細胞膜のフリッパーゼ活性を制御していることが示唆された.これらの結果は,KBM7細胞だけでなく,マウスTリンパ腫細胞株であるWR19L細胞,マウスpro B細胞株であるBa/F3細胞においても同様に再現された.以上より,ヒトやマウスの血球系細胞においてATP11Cが細胞膜で機能する主要なPtdSerフリッパーゼであること,ATP11C以外にもCDC50Aに依存して細胞膜で働くフリッパーゼが存在すること,そして,CDC50Aが細胞膜のPtdSerの非対称分布に必須であることが明らかとなった14, 18).
4. ATP8A2, ATP11AもPtdSerフリッパーゼ活性を持つ
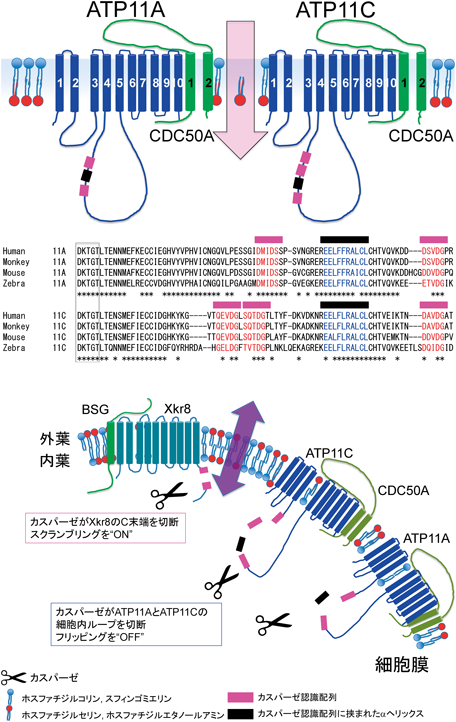
それでは,ATP11C以外にどのメンバーが細胞膜のPtdSerをフリップするのであろうか.ATP11Cを欠損させたWR19L細胞に,CDC50Aを安定発現させたのち,ヒトP4-ATPaseのファミリーメンバーを一つずつ発現させ,どのメンバーが細胞膜におけるフリッパーゼ活性を回復させるかをNBD-PSの取り込みで評価した19).その結果,ATP11Cの他にATP11AとATP8A2がNBD-PSの取り込みを強く回復させた.また,ATP11Cと同様に,ATP11AとATP8A2もCDC50A依存的に細胞膜に局在したことから,ATP8A2, ATP11A, ATP11Cの三つのメンバーが細胞膜で機能するCDC50A依存的なPtdSer-フリッパーゼであることが示唆された(表1).次に,これらの結果を生化学的に検証した19).HEK293T細胞にCDC50AとFLAGタグを付加したATP8A2, ATP11A, ATP11Cを発現させ,抗FLAG抗体を用いた免疫沈降法で精製した.各種タンパク質はCDC50Aと共沈されており,フリッパーゼ複合体として精製された.このフリッパーゼ複合体を,試験管内でリン脂質と反応させ,リン脂質応答性のATPase活性を評価した.その結果,フリッパーゼ複合体のATPase活性は低濃度のPtdSerとPtdEtnで強く上昇した(Ka=1~2 μM).一方,PtdChoやSMはATPase活性をまったく上昇させなかった.このATPase活性は,P型ATPaseの活性阻害剤としても機能するバナジン酸により抑制された.PtdSerとPtdEtnに特異的であること,バナジン酸感受性であることはこれまでに生体膜で検出された“flippase”活性の指標であった.以上,細胞を用いた実験と精製タンパク質を用いた生化学実験の結果から,ATP8A2, ATP11A, ATP11Cが哺乳類細胞の細胞膜で機能するフリッパーゼであると結論した.ATP8A2, ATP11A, ATP11Cが哺乳類細胞のフリッパーゼであるという結果は,国内外のグループでも追試されており,現在,ATP11AとATP11Cがヒト・マウスの組織に幅広く発現する主要な細胞膜フリッパーゼであると考えられている12).一方,ATP8A2はヒトとマウスともに神経細胞,網膜視細胞,精子など限られた細胞に発現し,また,ゴルジ体に局在するとの報告もあり12, 20),これらの細胞で特殊な役割を担う可能性がある.
表1 PtdSer-フリッパーゼ| クラス | α-サブユニット | β-サブユニット | 基質 | 細胞内局在 | 遺伝子発現 | カスパーゼ認識配列 | ヒトの疾患 | 遺伝子欠損マウス |
|---|
| 1a | ATP8A1 | CDC50A | PtdSer, PtdEtn | ゴルジ体,エンドソーム | 全身 | No | — | 学習障害 |
| ATP8A2 | CDC50A | PtdSer, PtdEtn | 細胞膜,ゴルジ体 | 脳,脊髄,網膜,精巣 | No | 精神遅滞,CAMRQ | 神経変性,網膜変性,難聴 |
| 6 | ATP11A | CDC50A | PtdSer, PtdEtn | 細胞膜 | 全身 | Yes (2) | — | 胎生致死 |
| ATP11B | CDC50A | PtdSer, PtdEtn | エンドソーム | 全身 | No | — | — |
| ATP11C | CDC50A | PtdSer, PtdEtn | 細胞膜 | 全身 | Yes (3) | 貧血 | 胆汁うっ滞,B細胞欠損,貧血 |
| PtdSerを基質とするP4-ATPaseファミリーメンバー.その他のメンバー(Class 1b, Class 2, Class 5)はPtdSerを基質としない.CAMRQ:小脳失調・精神遅滞および平衡障害症候群.—:不明.ATP11Bは,精製タンパク質を用いた実験で,アミノリン脂質依存的なATPase活性の上昇が検出されている45). |
細胞膜フリッパーゼはいずれもCDC50Aと結合することで安定なフリッパーゼ複合体を形成し,小胞体から細胞膜へ輸送される.しかし,CDC50Aがフリッパーゼの折りたたみをサポートする構造的なシャペロン分子としてのみ機能するのか,何らかの形でアミノリン脂質のフリッピングや基質特異性にも関与するのかについて明確な答えが得られていない.そこで,CDC50Aのどのアミノ酸残基がフリッパーゼの機能に重要であるかを網羅的に解析した21).ヒトCDC50AのcDNA配列にerror-prone PCR法を用いて平均2~3か所の変異をランダムに導入し,約1000万種類のヒトCDC50A変異体を作製した.この変異体のライブラリーを,レトロウイルスを用いて,ATP11Cを安定発現させたCDC50A欠損WR19L細胞に発現させた.次いで,感染細胞の細胞膜におけるフリッパーゼ活性を解析し,フリッパーゼ活性が上昇しない細胞(NBD-PSの取り込みが弱い細胞)をセルソーターで回収した.この細胞では,変異CDC50Aと複合体を形成したATP11Cが細胞膜でフリッパーゼとして機能できないと考えられた.回収した細胞を増殖させたのち,再度NBD-PSを用いたアッセイを行い,細胞膜のフリッパーゼ活性が回復しない細胞(Lost-flippase細胞)を濃縮した.続いて,Lost-flippase細胞に組み込まれたCDC50AのcDNA配列をPCR法で増幅し,増幅された配列を次世代シークエンサーで決定した.どのアミノ酸の変異がスクリーニングによって濃縮されたかを統計的に解析した結果,細胞外領域に位置する14か所のアミノ酸残基を同定した(Tyr82, Cys91, Cys94, Cys102, Cys104, Leu124, Asp181, Trp260, Lys271, Leu286, Tyr291, Val295, Tyr299, Lys308).同定したアミノ酸残基は,酵母からヒトまで広く保存されていた.実際,これらのアミノ酸に変異を導入したCDC50A変異体の大部分は,ATP11Cと安定的なフリッパーゼ複合体を形成できず,精製したATP11C-CDC50A変異体は凝集していた.さらに,この複合体をPtdSerと反応させてもATPase活性は上昇せず,フリッパーゼとして機能しないことが示された.また,これらのCDC50A変異体をCDC50A欠損細胞に発現させた場合,ATP11Cは小胞体から出ることができなかった.一方,260番目のトリプトファン残基(W260)に変異を導入したCDC50AをATP11Cとともに発現・精製すると,安定かつ凝集のないフリッパーゼ複合体を検出することができた.しかし,精製した複合体のPtdSerによるATPase活性の上昇は微弱なものであり,フリッパーゼとして十分に機能しないことが示唆された.実際,CDC50A-W260変異体を発現したWR19L細胞にATP11Cを発現させた場合,ATP11Cは細胞膜に局在するにもかかわらず,細胞膜でのフリッパーゼ活性は著しく低かった.この結果は,ATP11Aにおいても同様にみられ,CDC50Aの細胞外領域がフリッパーゼの折りたたみをサポートするだけでなく,細胞膜フリッパーゼの構造変化やPtdSerの移層にも何らかの形で関与する可能性を示唆した.今後,結晶構造解析やクライオ電子顕微鏡解析により,フリッパーゼがどのようにリン脂質を移層するのか,その移層機構を原子レベルで解明する必要がある.
アポトーシス細胞が“eat me”シグナルとしてPtdSerを露出する際,細胞膜においてリン脂質を双方向に輸送する活性であるスクランブラーゼ活性の上昇とフリッパーゼ活性の低下が起きることが報告されていた22, 23).しかし,その分子実体や活性制御のメカニズムは長らく不明であり,死細胞におけるPtdSer露出の分子機構は当分野の重要な問題であった.2013年,当時筆者らの研究室に所属していた鈴木淳博士らによって,アポトーシス時に活性化するスクランブラーゼXkr8が同定された24).Xkr8は生細胞において不活性体として細胞膜に局在するが,アポトーシス時にそのC末端がカスパーゼにより切断され,多量体化することでリン脂質のスクランブリングを活性化させてPtdSerを露出させる25).実際,Xkr8を欠損した血球系細胞株は,アポトーシス時にスクランブリングを起こすことができず,PtdSerを露出しない.それでは,フリッパーゼはどのように不活性化されているのであろうか.ATP11A, ATP11CおよびATP8A2のアポトーシス時の制御を解析した結果,ATP11AとATP11Cがカスパーゼにより速やかに切断・分解されることが明らかとなった(図4).ATP11AとATP11Cは,細胞質内ループの同じ場所に(種間で保存されたαヘリックスをはさむ形で),それぞれ2~3か所のカスパーゼ認識配列を有しており,この配列は高度に保存されていた.ATP11AやATP11Cのフリッパーゼ複合体はPtdSerとPtdEtnによるATPase活性の増大が検出されるが,野生型のATP11AやATP11Cをカスパーゼで処理するとそのATPase活性の増大は検出されなかった.一方,すべてのカスパーゼ認識配列に変異を導入した組換えタンパク質はカスパーゼ処理により切断されず,アミノリン脂質に応答してATPase活性を上昇させた19).興味深いことに,カスパーゼによるフリッパーゼの不活性化は,1か所のカスパーゼ認識配列の切断では不十分であった.すなわち,保存されたαヘリックスをはさんだ二つのカスパーゼ認識配列が両方とも切断され,間のαヘリックスがフリッパーゼ分子から完全に切り出されたときにATPase活性を失った.生化学実験の結果と同様に,カスパーゼ認識配列に変異を導入したATP11AやATP11Cを発現する細胞は,アポトーシス時にフリッパーゼが分解されず,PtdSerを露出しなかった14, 19).その結果,変異体発現細胞は,マクロファージに認識・貪食されなかった.以上より,カスパーゼによるATP11AおよびATP11Cの切断・不活性化は,アポトーシス時のPtdSerの露出とマクロファージによる貪食に必須であると結論した.アポトーシス細胞におけるPtdSer露出の分子機構は次のように想定される.アポトーシス細胞内で活性化したカスパーゼが,ATP11AとATP11Cの細胞質領域の機能ドメインを多重切断することで細胞膜のフリッパーゼ活性を消失させる.しかし,フリッパーゼの消失だけでは短時間にPtdSerが細胞膜の外葉に移層されない.そこで,カスパーゼがXkr8のC末端を切断することで多量体化を誘導し,細胞膜のスクランブリングを活性化させることで速やかにPtdSerを露出すると考えられる3, 4).カスパーゼによるフリッパーゼとスクランブラーゼの切断を介した活性制御は不可逆的な反応であり,アポトーシス細胞のPtdSerの露出が不可逆であることと一致する.ATP11AとATP11C, Xkr8は全身性に発現することから,多くのアポトーシス細胞において上記のプロセスを介したPtdSerの露出が起きていると考えられる.一方,ATP8A2はアポトーシス時に切断されなかった.ATP8A2はヒト,マウスともに神経細胞や精子などの限られた細胞に発現し,これらの細胞はアポトーシスを含めさまざまな局面でPtdSerを露出すると考えられている2).今後,ATP8A2がこれらの細胞のどの部分に局在し,どのような活性制御を受けているのか,またどのような機能を担っているのかを解析する必要があろう.
細胞内のカルシウム濃度が大きく上昇した細胞も,PtdSerを露出することが知られている.この局面においても,細胞膜においてスクランブラーゼ活性の上昇とフリッパーゼ活性の低下が起きると想定される26).カルシウム活性型のスクランブラーゼであるTMEM16Fは,2010年に鈴木らによる発現クローニング法によって同定された27).TMEMF16Fは細胞膜に局在する10回膜貫通タンパク質であり,ホモ二量体として存在する.膜貫通領域にカルシウムが直接結合すると,構造が変化し,リン脂質スクランブリングが活性化する.実際,各種リンパ球系の細胞にカルシウムイオノフォアを処理すると,カルシウムの上昇に応じた強いPtdSer露出がみられる.このPtdSer露出は,TMEM16F欠損細胞では検出されない.一方,カルシウムによるフリッパーゼの不活性化機構については明確な答えが得られていない.試験管内において,フリッパーゼ複合体のPtdSer依存的なATPase活性の上昇は,カルシウムによって濃度依存的に阻害される.しかし,その50%阻害濃度はATP8A2, ATP11A, ATP11Cでそれぞれ約500, 200, 100 μMであった19).サイトカインなどにより細胞が活性化される局面では,細胞膜近傍において,カルシウムの濃度が100 μM以上になることが報告されているが28),この濃度が細胞膜フリッパーゼの不活性化をすべて説明できるのかは不明である.また,カルシウムの上昇によりATP11Cがエンドサイトーシスされ,細胞膜から離れることでフリッパーゼ活性が抑制されることが報告されている29).しかし,ATP11Aはカルシウムの上昇に反応せずに細胞膜に局在し続けるとされており,細胞膜フリッパーゼに共通する阻害機構ではないと考えられる.いずれの阻害機構であっても,カスパーゼによるタンパク質分解とは異なり,カルシウムによるフリッパーゼの不活性化は可逆的である.したがって,露出したPtdSerは細胞内のカルシウムの減少に応じて,細胞膜フリッパーゼにより速やかに内葉へ戻されると考えられる(後述).実際,生体内ではさまざまな細胞がダイナミックにPtdSerの露出と取り込みを繰り返していることが報告されている2).いずれの局面においてもフリッパーゼとスクランブラーゼを制御する可逆的な機構が働いていると考えられ,今後,これらの制御機構を明らかにする必要があろう.
P4-ATPaseの中でも限られたメンバーのみが,細胞膜PtdSer-フリッパーゼとして機能する(表1).それでは,高等生物において細胞膜フリッパーゼはどのような生理機能を持つのであろうか.たとえば,ATP8A2欠損マウスは,神経変性,網膜変性,難聴の表現型を示し,ヒトにおいてもATP8A2の変異は,小脳失調・精神遅滞および平衡障害症候群(CAMRQ)の原因となる12).一方で,ATP11A欠損マウスは胎生致死となり(未発表),ATP11C欠損マウスは明らかになっているものだけでも,B細胞欠損,肝内胆汁うっ滞,貧血,難産など多様な症状を示すことが報告されている30–33).また,ヒトの貧血患者においてもATP11Cの変異が同定された34).しかし,細胞膜フリッパーゼの欠損や変異がどのようにこれらの病態を惹起するのか,そのメカニズムは不明であった.そこで,最も重篤な表現型の一つであるATP11C欠損マウスにおけるB細胞欠損症の分子メカニズムの解明を試みた35).2011年にオーストラリアとアメリカのグループが独立して,骨髄のpre B細胞,immature B細胞,mature B細胞,末梢血中のB細胞の数が野生型の数%程度にまで激減するマウスのラインとして,ATP11C欠損マウスを同定した30, 31).そこで,オーストラリアのグループで樹立されたATP11C欠損マウスを分与いただき,各種B細胞が既報のとおり減少していることを確認した(表2).ATP11AとATP11Cは全身性に発現し,細胞膜において同等のフリッパーゼ活性を持つ.そこで,野生型のマウス骨髄から各分化段階のB細胞をセルソーターで分取し,細胞膜フリッパーゼの遺伝子発現をRT-PCR法を用いて解析した.その結果,ATP11Cの遺伝子発現はどの分化段階のB細胞においても検出されたが,ATP11Aの遺伝子発現はpro B細胞以降の細胞で消失していた.つまり,ATP11C欠損マウスでは,pre-pro B細胞を除く大部分の前駆B細胞が両方のフリッパーゼを欠損した状態にあることが明らかとなった.そこで,ATP11A-ATP11C遺伝子二重欠損の影響を解析するため,CRISPR-Cas9法を用いてWR19L細胞のATP11AとATP11C遺伝子を破壊し,二重欠損細胞の細胞膜におけるPtdSerの動態を解析した.予想どおり,二重欠損細胞の細胞膜ではNBD-PSを取り込むフリッパーゼ活性がほぼ検出されなかった.しかし,意外なことに,通常状態におけるPtdSerの非対称的な分布は樹立されていた.この結果は,少なくともリンパ球系の細胞において,細胞膜のPtdSerの非対称性の樹立に細胞膜フリッパーゼが必須でないことを示唆した.そこで,細胞内のカルシウム濃度をカルシウムイオノフォアで上昇・活性化させ,PtdSerを露出させたあとの挙動を解析した.野生型の細胞は,イオノフォア処理によりPtdSerの露出が検出されたが,薬剤を培養系から除くことで速やかにPtdSerを細胞膜の内葉へ移層した.一方,変異細胞は,イオノフォアにより露出したPtdSerを反応収束後も露出し続けた.つまり,細胞膜フリッパーゼの機能は,刺激の収束に応じて露出したPtdSerを内葉に戻すこと,あるいは継続的なPtdSerの露出を防ぐことにあることが明らかとなった.この結果は,in vivoにおいても確認された.ATP11C欠損マウスの骨髄に残存する前駆B細胞は,著しく細胞膜フリッパーゼ活性が減少し,かつ40~50%の細胞が生きながらにPtdSerを露出していた.このPtdSerの露出は,野生型マウスから回収した前駆B細胞ではほぼ検出されなかった.
表2 骨髄におけるB細胞の分化 | pre-pro B細胞 | pro B細胞 | pre B細胞 | immature B細胞 | mature B細胞 |
|---|
| ATP11A | ○ | × | × | × | × |
| ATP11C | ○ | ○ | ○ | ○ | ○ |
| 野生型 | 3.9 | 3.0 | 17.0 | 7.2 | 17.0 |
| ATP11C欠損型 | 4.1 | 1.9 | 0.6 | 0.5 | 3.7 |
| MerTK-Axl二重欠損型 | 3.0 | 2.8 | 16.0 | 6.4 | 12.0 |
| MerTK-Axl-ATP11C三重欠損型 | 4.0 | 2.2 | 10.0 | 4.3 | 14.0 |
| (上)各B細胞の分化段階におけるATP11AとATP11Cの遺伝子発現.(下)大腿骨中のB細胞数の平均(×105/femur).ATP11C欠損マウスの骨髄ではpre B細胞以降の細胞数が減少しているが(太字,赤色),MerTK-Axl二重欠損の遺伝背景においてはATP11C欠損によるB細胞の減少がみられない. |
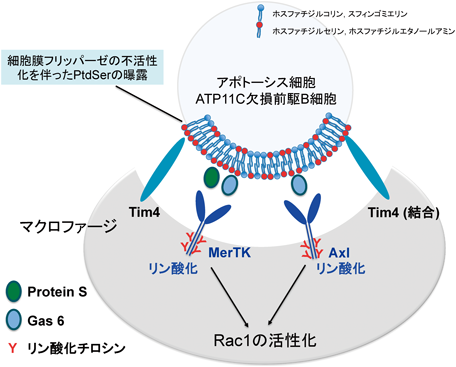
PtdSerはマクロファージへの“eat me”シグナルとして機能し,アポトーシス細胞は生体で速やかに除去される.したがって,通常,生体内の組織に残存するアポトーシス細胞は検出されない.この知見は,ATP11Cを欠損したB細胞が骨髄中でPtdSer依存的にマクロファージに貪食されること,この貪食がB細胞欠損の原因である可能性を示唆した.マクロファージは,種々のPtdSer結合分子を用いて死細胞を貪食する3)(後述).たとえば,腹腔常在性マクロファージは1型膜タンパク質のPtdSer受容体であるTim4と,受容体型チロシンキナーゼであるMerTKやAxlを強く発現し,Tim4で死細胞をマクロファージに結合させ,MerTKやAxlを介したチロシンリン酸化シグナルを効率よく伝達することで死細胞を貪食する36, 37).同様に,骨髄にもTim4およびMerTKとAxlを強く発現する常在性マクロファージが多く存在した.そこで,MerTKやAxlのチロシンキナーゼ活性阻害剤であるBMS-777607をATP11C欠損マウス腹腔に1日に2回,計4日間注射し,B細胞欠損症が改善するかを検討した.その結果,ATP11C欠損マウスの骨髄におけるpre B細胞とimmature B細胞の減少が有意に改善し,野生型と同程度の数に回復することが明らかとなった.この結果を,遺伝子改変マウスでも確認した.ATP11C欠損マウスとMerTK欠損マウス,Axl欠損マウス,あるいはMerTK-Axl二重欠損マウスとを交配させることでATP11CとMerTK-Axlの多重欠損マウスを作製し,これらのマウスの骨髄のB細胞を解析した.その結果,ATP11C遺伝子欠損による骨髄のB細胞数の減少は,MerTKとAxlの遺伝子欠損で段階的に回復し,MerTK-Axlの二重欠損により野生型と同程度にまで回復することが明らかとなった(表2).MerTK-Axl-ATP11C三重欠損マウスの骨髄から回収したpro B細胞,pre B細胞,およびimmature B細胞の40~50%がPtdSerを露出しており,骨髄内でマクロファージに貪食されないことで細胞数が回復したと考えられた.実際,三重欠損マウスの骨髄から単離した前駆B細胞は,培養ディッシュ上で腹腔常在性マクロファージにPtdSer依存的に貪食されること,この貪食はMerTK-Axlを二重欠損したマクロファージではみられないことを確認した.また,ATP11C欠損マウスの骨髄中には,貪食した細胞の分解産物を含むマクロファージが多く存在しており,活発にB細胞を貪食していることが示唆された(順天堂大学内山安男博士との共同研究).以上より,骨髄に存在するATP11C欠損前駆B細胞が一過的に露出したPtdSerを内葉へ戻すことができず,マクロファージにPtdSer依存的に貪食されることがB細胞欠損症の原因であると結論した.
マクロファージは生きた細胞を貪食することはなく,アポトーシス細胞を特異的に貪食する.この“生細胞とアポトーシス細胞の識別”には,PtdSerが重要な役割を担う.1983年に,PtdSerを赤血球の細胞膜に人工的に取り込ませ,PtdSerを露出させた赤血球がマクロファージに貪食されることが報告された38).その後,アポトーシス細胞がPtdSerを露出すること,露出されたPtdSerをPtdSer結合タンパク質でマスクした場合や,PtdSerを含むリポソームで競合させた場合にはマクロファージがアポトーシス細胞を貪食できないことが報告された39).アポトーシス細胞によるPtdSerの露出やPtdSerを介したアポトーシス細胞の貪食は,線虫からヒトまで保存されており,多細胞生物に広く保存された生命現象の一つである.その後,特にヒト,マウスなどの高等動物において,マクロファージがどのようにPtdSerを認識しているのか,PtdSerの認識と貪食機構の研究が国内外でなされた.これまでに,PtdSerの認識には,分泌タンパク質を介した系と膜タンパク質を介した系の二つのシステムが存在することが明らかになっている3, 40).分泌型のPtdSer結合タンパク質として,MFG-E8, Protein S, Gas 6の三つのタンパク質が同定されている.MFG-E8は糖鎖を持つ46 kDaのタンパク質であり,そのC末端に存在するFactor VIII相同領域が死細胞のPtdSerと結合し,同時にN末端に存在するRGDモチーフが貪食細胞に発現するIntegrin-αvβ3のリガンドとして作用する.すなわち,アポトーシス細胞とマクロファージの“橋渡し”の形をとることで,死細胞の貪食を促進する.“橋渡し”のシステムは,Protein SとGas 6においてもみられる.Protein SとGas 6はアミノ酸配列で約40%一致する血清タンパク質であり,そのN末端にあるGLA領域がPtdSerと結合し,C末端の性ホルモン結合性グロブリン領域がマクロファージに発現するTAM(Tyro3, Axl, Mer)受容体型チロシンキナーゼのリガンドとして機能することで死細胞の貪食を促進する.一方,PtdSerに直接結合する膜タンパク質(PtdSer受容体)としてTim4が同定されている36).Tim4は,1型膜タンパク質であり,腹腔常在性マクロファージをはじめ,骨髄,肝臓,脾臓,腸管などさまざまな組織に常在するマクロファージに特異的に発現する41).Tim4の細胞外領域にあるIgV領域がPtdSerと強固に結合し,アポトーシス細胞をマクロファージの細胞膜に直接結合させることで貪食を促進する.筆者らは,貪食能をまったく持たないBa/F3細胞を用いて,アポトーシス細胞の貪食の再構成を試みた37).Ba/F3にTim4を単独で発現させると,アポトーシス細胞を表面に結合させるが貪食の効率は低いままであった.同様に,MerTKを単独で発現させた場合は,アポトーシス細胞を結合させることができずに貪食することができなかった.一方,Tim4とMerTKを共発現させたBa/F3細胞は,アポトーシス細胞を効率よく結合・貪食し,マクロファージに匹敵する貪食能を示した.同様の結果は,マウス腹腔常在性マクロファージでも得られた.Tim4欠損マウスから調製した腹腔常在性マクロファージは,アポトーシス細胞を表面に結合することがまったくできない.その結果,MerTKのチロシンリン酸化が誘導できず,アポトーシス細胞を貪食することができなかった.一方で,MerTK欠損マウスの腹腔常在性マクロファージは,アポトーシス細胞を強固に結合させることができるが,MerTKを介したチロシンリン酸化シグナルを伝達することができず,貪食することができなかった.これらの結果は,Tim4によって死細胞を食細胞の表面に結合させるプロセスが,MerTKのシグナル伝達に重要な役割を担うことを示唆した.そこで,NIH3T3細胞を用いて,MerTKが死細胞の貪食を惹起するのに必要なリガンド濃度を解析した.その結果,Tim4を発現させることにより,貪食を誘導するのに必要なProtein SやGas 6の濃度が数十分の一に低下することが明らかとなった41).まとめると,腹腔常在性マクロファージは,PtdSer受容体であるTim4を用いてアポトーシス細胞を細胞表面に結合させる(Binding step),この結合によって,TAM受容体であるMerTKの貪食シグナルが効率よく伝達される(Tickling step).この二つのステップが相乗的に作用することで,PtdSer依存的なアポトーシス細胞の貪食が遂行されると結論した(図5).生体にはTim4とTAM受容体を共発現するマクロファージが多く存在し,それらのマクロファージも同様の機構で死細胞を貪食すると考えられる.
上述のとおり,マクロファージはさまざまなPtdSer結合タンパク質を用いてアポトーシス細胞を特異的に認識して貪食する.しかし,アポトーシス細胞のPtdSerが単独でマクロファージの貪食を誘導できるのか,あるいはアポトーシスに関連する他のシグナルも必要とするのかという問題については明確な答えが得られていなかった.すなわち,“貪食の十分条件”問題である.過去に,PtdSerを人工的に細胞膜に大量に外挿した細胞がマクロファージに貪食されるという結果が得られていたが,人工的な系であり,どの程度の妥当性があるのか不明であった.また,生きた細胞は“don’t eat me”シグナルを構成的に提示することで,マクロファージによる貪食を防いでいるという仮説も提唱されており,混沌とした状況であった.この問題に明確に答えるには,アポトーシス細胞と同程度のPtdSerを定常状態で露出する“生きた細胞”がマクロファージに貪食されるかを解析する必要があったが,そのような哺乳類細胞は存在しなかった.筆者らは,フリッパーゼとスクランブラーゼの研究の過程でPtdSerを構成的に露出する二つのタイプのリンパ球系細胞を取得することに成功した.一つは,カルシウム活性型スクランブラーゼであるTMEM16Fのconstitutive active変異体を発現したWR19L細胞である.この変異体発現細胞は,細胞膜での強いリン脂質スクランブリングが恒常的に起きることによりPtdSerを生きながらに細胞表面に露出する.一方,CDC50Aを欠損したWR19L細胞は,フリッパーゼ活性が完全に消失することで構成的に細胞膜の外葉にPtdSerを露出する.これらの細胞をマクロファージと共培養した結果,CDC50Aを欠損した細胞は生きたままPtdSer依存的に貪食されたのに対し,TMEM16F変異体を発現させた細胞は貪食されなかった14, 42).TMEM16F変異体を発現した細胞では,PtdSerがTMEM16FによるスクランブリングとATP11A/ATP11Cによるフリッピングの両方によって移層されることで,脂質二重層間を速い速度で移動していると考えられる.細胞表面に露出したPtdSerは頻繁に内葉へ戻されることから,PtdSer結合タンパク質とPtdSerとの強固な結合が短時間に成立しづらく,貪食に至らないと考えられる4).一方,CDC50A欠損細胞は,フリッパーゼ活性を失うことでPtdSerが細胞表面に安定的に露出される.その結果,PtdSerがPtdSer結合タンパク質と相互作用することができ,マクロファージに貪食シグナルが伝達されると考えられた.アポトーシス細胞においてもフリッパーゼは不活性化され,内在性のスクランブラーゼの分子数に応じた生理的な範囲のスクランブリングが起こることから,PtdSerが安定的に露出されると考えられる.以上より,フリッパーゼ活性の消失を伴ったPtdSerの安定的な露出は,マクロファージの貪食を十分に誘導できると結論した.先述のとおり,ATP11C欠損マウスの骨髄では,前駆B細胞がマクロファージに活発に貪食されており,生体内においてもPtdSerが貪食の十分条件として機能すると考えられる.WR19L細胞やマウス前駆B細胞は,“don’t eat me”シグナルの候補であるCD47分子を強く発現している.PtdSerを露出したこれらの細胞が生きながらにマクロファージに貪食されるという実験結果は,少なくともPtdSerがCD47の発現にかかわらず“eat me”シグナルとして機能することを示唆した.CD47はImmune inhibitory receptorであるSIRPαのリガンドとして機能し,SIRPαを発現する免疫細胞に抑制性のシグナルを伝達する.CD47-SIRPαの経路は“don’t eat me”シグナルと提唱されているが,今後,CD47-SIRPα経路の阻害だけでマクロファージの貪食を十分に誘導できるのか,他の貪食シグナルが必要であるのか,あるいはCD47-SIRPα経路がマクロファージを“calm down”させているのか,について明確な答えが出ることが望まれる.
一連の研究によりATP11AとATP11Cがヒトやマウスの細胞に幅広く発現する細胞膜フリッパーゼであることが明らかとなった.アポトーシスを起こした細胞は,フリッパーゼを不可逆的に切断・分解し,PtdSerを安定的に露出することでマクロファージに貪食される.死細胞の運命を一方向かつ不可逆的に決定づける上で,カスパーゼによるフリッパーゼの不活性化とスクランブラーゼの活性化を介したPtdSerの露出は,精巧かつ頑強なシステムである.一方,細胞膜フリッパーゼは,通常状態におけるPtdSerの非対称的分布の樹立には必須でなかった.この結果は,ATP11AとATP11Cに相当するオルソログを持たない酵母においても,細胞膜のPtdSerの非対称性が樹立されることと一致する.PtdSerの非対称性の樹立は,細胞内のオルガネラにおけるPtdSerの分布から考える必要があるだろう.それでは,ATP11AとATP11Cは何のために存在するのであろうか.ATP11C欠損マウスの解析により,細胞膜フリッパーゼの生理機能の一つは,露出したPtdSerを刺激の収束に応じて“速やかに”内葉に移層し,マクロファージの誤認識による貪食を防ぐことであることが明らかとなった.では,なぜマクロファージに貪食される危険がありながらPtdSerを露出する必要があるのであろうか.生体では,さまざまな細胞が一過的にPtdSerを露出していると考えられ,前駆B細胞,肥満細胞,筋肉細胞,活性化した精子,神経細胞,マクロファージ,あるいはがん組織でみられる血管内皮細胞などが報告されている2).今後,これらの局面で機能するスクランブラーゼを明らかにし,その遺伝子欠損マウスを解析することでPtdSerの持つ新たな生理機能を明らかにする必要があろう.また,細胞膜フリッパーゼには,マクロファージによる貪食を防ぐ以外にも機能があると考えられる.実際,ATP11C欠損マウスの貧血や肝内胆汁うっ滞の表現型は,MerTK-Axl二重欠損の遺伝背景においても改善しなかった.過去に,フリッパーゼが細胞膜の構造や流動性などの物性を変化させることが報告されている43).実際,ATP11Cを欠損した赤血球の細胞膜は異常な形態を示し,寿命が短くなる32).また,ATP11Cを欠損した肝細胞では,細胞膜上の胆汁トランスポーターの発現が著しく減少する44).フリッパーゼ活性の減少がどのように細胞膜の物性を変化させるのか,その変化がどのように赤血球や肝細胞の細胞膜の機能や膜タンパク質の挙動に影響を与えるのか,そのメカニズムを明らかにする必要があろう.細胞膜のリン脂質を介した細胞間コミュニケーションや生理機能にはまだまだ多くの謎が残されており,今後の進展が期待される.
謝辞Acknowledgments
本稿では,筆者が長田重一教授の研究室で博士研究員として研究を始めてから現在までに得られた知見について概説しました.長きにわたりご指導いただいた長田重一先生に,心より感謝申し上げます.また,研究室の先輩,同僚,後輩,学生さん,秘書さん,技官さん,共同研究者の先生,多くの方々にサポートいただきました.重ねて御礼申し上げます.
引用文献References
1) van Meer, G., Voelker, D.R., & Feigenson, G.W. (2008) Membrane lipids: Where they are and how they behave. Nat. Rev. Mol. Cell Biol., 9, 112–124.
2) Bevers, E.M. & Williamson, P.L. (2016) Getting to the outer leaflet: Physiology of phosphatidylserine exposure at the plasma membrane. Physiol. Rev., 96, 605–645.
3) Nagata, S. (2018) Apoptosis and clearance of apoptotic cells. Annu. Rev. Immunol., 36, 489–517.
4) Segawa, K. & Nagata, S. (2015) An apoptotic ‘Eat me’ signal: phosphatidylserine exposure. Trends Cell Biol., 25, 639–650.
5) Yoshida, H., Kawane, K., Koike, M., Mori, Y., Uchiyama, Y., & Nagata, S. (2005) Phosphatidylserine-dependent engulfment by macrophages of nuclei from erythroid precursor cells. Nature, 437, 754–758.
6) Schroit, A.J., Madsen, J.W., & Tanaka, Y. (1985) In vivo recognition and clearance of red blood cells containing phosphatidylserine in their plasma membranes. J. Biol. Chem., 260, 5131–5138.
7) Pomorski, T.G. & Menon, A.K. (2016) Lipid somersaults: Uncovering the mechanisms of protein-mediated lipid flipping. Prog. Lipid Res., 64, 69–84.
8) Tanaka, K., Fujimura-Kamada, K., & Yamamoto, T. (2011) Functions of phospholipid flippases. J. Biochem., 149, 131–143.
9) Toyoshima, C. (2009) How Ca2+-ATPase pumps ions across the sarcoplasmic reticulum membrane. Biochim. Biophys. Acta, 1793, 941–946.
10) Saito, K., Fujimura-Kamada, K., Furuta, N., Kato, U., Umeda, M., & Tanaka, K. (2004) Cdc50p, a protein required for polarized growth, associates with the Drs2p P-type ATPase implicated in phospholipid translocation in Saccharomyces cerevisiae. Mol. Biol. Cell, 15, 3418–3432.
11) Kato, U., Emoto, K., Fredriksson, C., Nakamura, H., Ohta, A., Kobayashi, T., Murakami-Murofushi, K., Kobayashi, T., & Umeda, M. (2002) A novel membrane protein, Ros3p, is required for phospholipid translocation across the plasma membrane in Saccharomyces cerevisiae. J. Biol. Chem., 277, 37855–37862.
12) Andersen, J.P., Vestergaard, A.L., Mikkelsen, S.A., Mogensen, L.S., Chalat, M., & Molday, R.S. (2016) P4-ATPases as phospholipid flippases-structure, function, and enigmas. Front. Physiol., 7, 275.
13) Baldridge, R.D. & Graham, T.R. (2012) Identification of residues defining phospholipid flippase substrate specificity of type IV P-type ATPases. Proc. Natl. Acad. Sci. USA, 109, E290–E298.
14) Segawa, K., Kurata, S., Yanagihashi, Y., Brummelkamp, T.R., Matsuda, F., & Nagata, S. (2014) Caspase-mediated cleavage of phospholipid flippase for apoptotic phosphatidylserine exposure. Science, 344, 1164–1168.
15) Carette, J.E., Guimaraes, C.P., Varadarajan, M., Park, A.S., Wuethrich, I., Godarova, A., Kotecki, M., Cochran, B.H., Spooner, E., Ploegh, H.L., et al. (2009) Haploid genetic screens in human cells identify host factors used by pathogens. Science, 326, 1231–1235.
16) Kotecki, M., Reddy, P.S., & Cochran, B.H. (1999) Isolation and characterization of a near-haploid human cell line. Exp. Cell Res., 252, 273–280.
17) Takatsu, H., Baba, K., Shima, T., Umino, H., Kato, U., Umeda, M., Nakayama, K., & Shin, H.W. (2011) ATP9B, a P4-ATPase (a putative aminophospholipid translocase), localizes to the trans-Golgi network in a CDC50 protein-independent manner. J. Biol. Chem., 286, 38159–38167.
18) Segawa, K., Suzuki, J., & Nagata, S. (2014) Flippases and scramblases in the plasma membrane. Cell Cycle, 13, 2990–2991.
19) Segawa, K., Kurata, S., & Nagata, S. (2016) Human Type IV P-type ATPases that work as plasma membrane phospholipid flippases and their regulation by caspase and calcium. J. Biol. Chem., 291, 762–772.
20) Coleman, J.A., Kwok, M.C., & Molday, R.S. (2009) Localization, purification, and functional reconstitution of the P4-ATPase Atp8a2, a phosphatidylserine flippase in photoreceptor disc membranes. J. Biol. Chem., 284, 32670–32679.
21) Segawa, K., Kurata, S., & Nagata, S. (2018) The CDC50A extracellular domain is required for forming a functional complex with and chaperoning phospholipid flippases to the plasma membrane. J. Biol. Chem., 293, 2172–2182.
22) Verhoven, B., Schlegel, R.A., & Williamson, P. (1995) Mechanisms of phosphatidylserine exposure, a phagocyte recognition signal, on apoptotic T lymphocytes. J. Exp. Med., 182, 1597–1601.
23) Bratton, D.L., Fadok, V.A., Richter, D.A., Kailey, J.M., Guthrie, L.A., & Henson, P.M. (1997) Appearance of phosphatidylserine on apoptotic cells requires calcium-mediated nonspecific flip-flop and is enhanced by loss of the aminophospholipid translocase. J. Biol. Chem., 272, 26159–26165.
24) Suzuki, J., Denning, D.P., Imanishi, E., Horvitz, H.R., & Nagata, S. (2013) Xk-related protein 8 and CED-8 promote phosphatidylserine exposure in apoptotic cells. Science, 341, 403–406.
25) Suzuki, J., Imanishi, E., & Nagata, S. (2016) Xkr8 phospholipid scrambling complex in apoptotic phosphatidylserine exposure. Proc. Natl. Acad. Sci. USA, 113, 9509–9514.
26) Bitbol, M., Fellmann, P., Zachowski, A., & Devaux, P.F. (1987) Ion regulation of phosphatidylserine and phosphatidylethanolamine outside-inside translocation in human erythrocytes. Biochim. Biophys. Acta, 904, 268–282.
27) Suzuki, J., Umeda, M., Sims, P.J., & Nagata, S. (2010) Calcium-dependent phospholipid scrambling by TMEM16F. Nature, 468, 834–838.
28) Marsault, R., Murgia, M., Pozzan, T., & Rizzuto, R. (1997) Domains of high Ca2+ beneath the plasma membrane of living A7r5 cells. EMBO J., 16, 1575–1581.
29) Takatsu, H., Takayama, M., Naito, T., Takada, N., Tsumagari, K., Ishihama, Y., Nakayama, K., & Shin, H.W. (2017) Phospholipid flippase ATP11C is endocytosed and downregulated following Ca(2+)-mediated protein kinase C activation. Nat. Commun., 8, 1423.
30) Yabas, M., Teh, C.E., Frankenreiter, S., Lal, D., Roots, C.M., Whittle, B., Andrews, D.T., Zhang, Y., Teoh, N.C., Sprent, J., et al. (2011) ATP11C is critical for the internalization of phosphatidylserine and differentiation of B lymphocytes. Nat. Immunol., 12, 441–449.
31) Siggs, O.M., Arnold, C.N., Huber, C., Pirie, E., Xia, Y., Lin, P., Nemazee, D., & Beutler, B. (2011) The P4-type ATPase ATP11C is essential for B lymphopoiesis in adult bone marrow. Nat. Immunol., 12, 434–440.
32) Yabas, M., Coupland, L.A., Cromer, D., Winterberg, M., Teoh, N.C., D’Rozario, J., Kirk, K., Bröer, S., Parish, C.R., & Enders, A. (2014) Mice deficient in the putative phospholipid flippase ATP11C exhibit altered erythrocyte shape, anemia, and reduced erythrocyte life span. J. Biol. Chem., 289, 19531–19537.
33) Siggs, O.M., Schnabl, B., Webb, B., & Beutler, B. (2011) X-linked cholestasis in mouse due to mutations of the P4-ATPase ATP11C. Proc. Natl. Acad. Sci. USA, 108, 7890–7895.
34) Arashiki, N., Takakuwa, Y., Mohandas, N., Hale, J., Yoshida, K., Ogura, H., Utsugisawa, T., Ohga, S., Miyano, S., Ogawa, S., et al. (2016) ATP11C is a major flippase in human erythrocytes and its defect causes congenital hemolytic anemia. Haematologica, 101, 559–565.
35) Segawa, K., Yanagihashi, Y., Yamada, K., Suzuki, C., Uchiyama, Y., & Nagata, S. (2018) Phospholipid flippases enable precursor B cells to flee engulfment by macrophages. Proc. Natl. Acad. Sci. USA, 115, 12212–12217.
36) Miyanishi, M., Tada, K., Koike, M., Uchiyama, Y., Kitamura, T., & Nagata, S. (2007) Identification of Tim4 as a phosphatidylserine receptor. Nature, 450, 435–439.
37) Nishi, C., Toda, S., Segawa, K., & Nagata, S. (2014) Tim4- and MerTK-mediated engulfment of apoptotic cells by mouse resident peritoneal macrophages. Mol. Cell. Biol., 34, 1512–1520.
38) Tanaka, Y. & Schroit, A.J. (1983) Insertion of fluorescent phosphatidylserine into the plasma membrane of red blood cells. Recognition by autologous macrophages. J. Biol. Chem., 258, 11335–11343.
39) Fadok, V.A., Voelker, D.R., Campbell, P.A., Cohen, J.J., Bratton, D.L., & Henson, P.M. (1992) Exposure of phosphatidylserine on the surface of apoptotic lymphocytes triggers specific recognition and removal by macrophages. J. Immunol., 148, 2207–2216.
40) Lemke, G. (2019) How macrophages deal with death. Nat. Rev. Immunol., 19, 539–549.
41) Yanagihashi, Y., Segawa, K., Maeda, R., Nabeshima, Y.I., & Nagata, S. (2017) Mouse macrophages show different requirements for phosphatidylserine receptor Tim4 in efferocytosis. Proc. Natl. Acad. Sci. USA, 114, 8800–8805.
42) Segawa, K., Suzuki, J., & Nagata, S. (2011) Constitutive exposure of phosphatidylserine on viable cells. Proc. Natl. Acad. Sci. USA, 108, 19246–19251.
43) Devaux, P.F., Herrmann, A., Ohlwein, N., & Kozlov, M.M. (2008) How lipid flippases can modulate membrane structure. Biochim. Biophys. Acta, 1778, 1591–1600.
44) Matsuzaka, Y., Hayashi, H., & Kusuhara, H. (2015) Impaired hepatic uptake by organic anion-transporting polypeptides Is associated with hyperbilirubinemia and hypercholanemia in Atp11c mutant mice. Mol. Pharmacol., 88, 1085–1092.
45) Wang, J., Molday, L.L., Hii, T., Coleman, J.A., Wen, T., Andersen, J.P., & Molday, R.S. (2018) Proteomic analysis and functional characterization of P4-ATPase phospholipid flippases from murine tissues. Sci. Rep., 8, 10795.
著者紹介Author Profile
 瀬川 勝盛(せがわ かつもり)
瀬川 勝盛(せがわ かつもり)大阪大学免疫学フロンティア研究センター免疫・生化学部門准教授.博士(理学).
略歴奈良県大和郡山市生まれ.奈良高校卒業.大阪府立大学工学部卒業,2009年大阪大学大学院生命機能研究科博士一貫課程修了(内分泌・代謝内科学講座),同年より京都大学大学院医学研究科博士研究員(長田重一教授),11年同助教を経て17年より大阪大学免疫学フロンティア研究センター免疫・生化学部門准教授.
研究テーマと抱負「細胞は何のために,どのように生体膜のリン脂質を動かしているのか?」についての研究.ヒトの疾患に関わる新しい分野にチャレンジしたい.
ウェブサイトhttp://biochemi.ifrec.osaka-u.ac.jp
趣味読書,サーフィン.