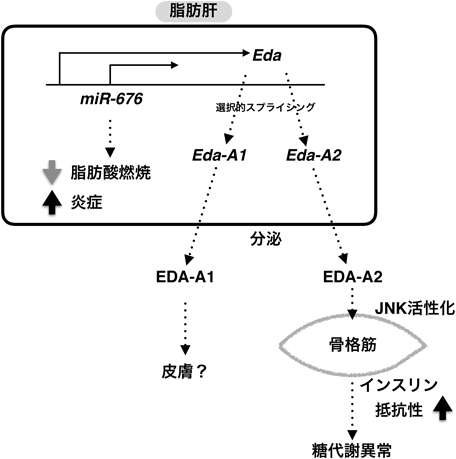
microRNAによる新規インスリン抵抗性惹起作用の解明An intronic-microRNA screening revealed a previously unknown mechanism of obesity-related insulin resistance
国立国際医療研究センター研究所Research Institute, National Center for Global Health and Medicine ◇ 〒162–8655 東京都新宿区戸山1–21–1 ◇ 1–21–1 Toyama, Shinjuku-ku, Tokyo 162–8655, Japan
発行日:2019年12月25日Published: December 25, 2019