ユビキチン鎖修飾を受けた基質タンパク質がプロテアソーム(プロテアーゼ複合体)によって認識・分解されるユビキチン–プロテアソームシステムの発見以降,細胞内の多くのタンパク質のターンオーバーを規定する必須因子としてユビキチンの重要性は確立された1).現在では,ユビキチンが関与する細胞内生理機能は拡大の一途をたどっており,細胞周期,DNA修復,細胞外刺激に応答するシグナル伝達など多岐に及ぶことが知られている.
しかしながら,ユビキチンがモノユビキチン化(一つのユビキチンが付加)やポリユビキチン化(複数のユビキチンがタンデムに連結し付加)といった修飾形態を駆使して,いかに多種多様な生命現象に関与するのか,その詳細な原理やメカニズムはいまだ解明されていない部分が多い.近年注目されているユビキチンコードは,ユビキチン鎖のタイプに注目した概念で,特定のタイプのユビキチン鎖を生成するユビキチン結合酵素(E3)とその鎖を認識可能なユビキチン鎖結合ドメイン(UBD)を有するデコーダータンパク質が存在することで以降発動される生理機能が規定されるという,一見しただけでは区別できないユビキチン鎖が予想以上に複雑な生命暗号を含んでいるという新しい考え方であり,E3による基質選択性やE3の活性制御機構と並んで,ユビキチン修飾システムの理解には欠かせない概念である2).
本稿で取り扱う直鎖状ユビキチン鎖は,大変興味深いことに我々が研究対象としているLUBAC(linear ubiquitin assembly complex)以外のE3では生成することができず,細胞内の直鎖状ユビキチン鎖量もユビキチン鎖全体の0.1%ほどしか存在しないにもかかわらず,NF-κBや細胞死制御といった細胞生存や恒常性維持に重要なシグナル伝達系に関与している.また,LUBACの構成因子をコードするHoipやHoil-1l遺伝子の欠損マウスが胎生致死に至ることから,個体の発生過程においても必要不可欠である3).他のユビキチン鎖では代替不可能な重要かつ特異的な生理機能を担う直鎖状ユビキチン鎖とLUBACの解析は,今後ユビキチンコードの直接的な理解に多大な貢献をしていくと確信している.
本稿では,LUBACを構成する各サブユニットの役割や,近年のLUBAC複合体の構造解析から明らかにされてきた直鎖状ユビキチン鎖形成機構の詳細について解説する.また,従来詳細に解析されてきたLUBACによるNF-κBシグナル経路の活性化メカニズムに加えて,最近筆者らはLUBACが細胞死と免疫機能の制御を介して自己炎症の発症を抑制していることを報告しており,簡単に解説する.
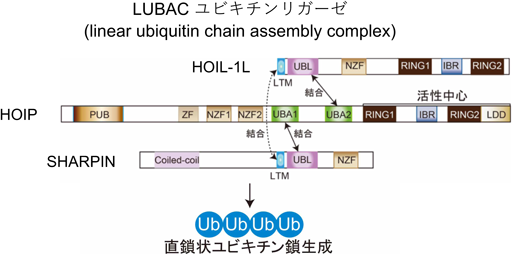
LUBACは,E3リガーゼ活性を持つサブユニットHOIP(HOIL-1L-interacting protein)(120 kDa)と,複合体の安定化に寄与するサブユニットHOIL-1L(heme-oxidized IRP2 ligase 1L)(58 kDa)とSHARPIN(SHANK-associated RH domain-interacting protein)(40 kDa)の三者からなるE3複合体酵素である(図1).細胞内では約600 kDaの高分子複合体として存在しており,その中には複数のLUBACが含まれていると想定されているが,存在比率や全体構造はいまだ明らかとされていない4, 5).これらLUBACを構成する三者のタンパク質は配列中にマルチドメインを有しており,LUBAC複合体の安定化や直鎖状ユビキチン鎖形成に関与する.
HOIPはRNF31(RING finger protein 31)とも呼ばれ,LUBACの直鎖状ユビキチン鎖修飾活性を担う触媒サブユニットである.LUBACはHOIPのC末端側のRING1-IBR(in-between-RING)-RING2(RBR)領域を介してリガーゼ活性を発揮することから,RBR型E3ファミリーに属する.HOIP中央部には,他のLUBAC構成サブユニットと複合体を形成するために必要とされるUBA(ubiquitin-associated)ドメインを有しており,UBA内N末端側のUBA1はSHARPINのUBL(ubiquitin-like)ドメインと,C末端側のUBA2はHOIL-1LのUBLと会合することで安定的な複合体の形成を可能としている6).UBA1のさらにN末端側には,ZF-NZF(Npl4 zinc finger)1-NZF2領域があり,基質であるNEMO(NF-κB essential modulator, IKKγ)とユビキチン鎖を認識する7).HOIPの最もN末端側にはPUB(peptide:N-glycanase/UBA or UBX-containing proteins)ドメインが存在する.PUBはAAA+ ATPaseであるp97/VCPと結合するアミノ酸配列としてPNGaseのN末端領域で見いだされたドメインであるが,現在では直鎖状ユビキチン鎖を特異的に切断する脱ユビキチン化タンパク質(DUB)OTULIN内のPIM(PUB interacting motif)ドメインや,同じくDUBであるCYLDとの会合を介在するアダプタータンパク質SPATA2(spermatogenesis-associated protein 2)内PIMとの結合領域として機能することが知られている8, 9).PIM領域内によく保存されたTyr残基がLUBACとの会合に必須であり,Tyrがリン酸化を受けた場合,LUBACとの結合が抑制されることが示されており,細胞内のリン酸化依存的シグナルの上昇がLUBACの直鎖状ユビキチン鎖形成を亢進させると予想されるが,その生理学的重要性は明らかにされていない.
HOIL-1LもまたHOIPと同じくC末端領域にRBRドメインを持つためE3に属するが,そのリガーゼ活性はきわめて低いため解析が困難であった.最近の報告ではHOIL-1Lのモノユビキチン化活性がToll-like受容体シグナル関連因子IRAK1, IRAK2およびMyD88のポリユビキチン化のイニシエーションに必須であることや,HOIL-1L自身やSHARPINのモノユビキチン化を行うことが示されている(イムノブロット解析においてLUBAC複合体中のこれらのタンパク質がダブレットバンドとして検出される原因と考えられる).さらに,HOIL-1Lは他のE3とは異なり,基質タンパク質上のセリン・トレオニン残基にエステル結合を介してユビキチンを付加することが報告されている10).HOIL-1LのNZF領域は直鎖状ユビキチン鎖と特異的に結合することが知られている.結晶構造の詳細な解析から,一般的なNZF-coreと呼ばれる領域を介して遠位ユビキチンの疎水性パッチ領域との相互作用に加え,NZF-tailと呼ばれるNZF C末端側α-ヘリックス領域を介して,近位ユビキチンとの結合親和性を亢進させ,直鎖状ユビキチン鎖への特異性を高めていることが示されている11).HOIL-1LのN末端にはLUBAC複合体を形成するために必要な領域が存在する.HOIPのUBA2と結合するUBLが配置され,さらには最近筆者らの研究室からLTM(LUBAC-tethering motif)と呼ばれるLUBAC複合体の安定化に寄与する新たな配列が見いだされ,SHARPINのLTMと結合することが示されている6).マウスレベルの解析から,Hoipと同様にHoil-1lを欠損したマウスでは耐性致死となることから,HOIL-1LがLUBAC複合体を規定する未知の役割を担っていることが予想される12).
もう一方のアクセサリー分子SHARPINのC末端領域は,HOIL-1LのN末端領域(LTM-UBL-NZF)と相同性が高い.SHARPINのLTMとUBLは,HOIL-1LのLTMとHOIPのUBA1それぞれと結合し,LUBAC複合体の安定化を担っている.NZFについては,筆者らの研究室からHOIL-1LのNZFとは機能的に異なることを報告しており,HOIL-1LのNZFが直鎖状ユビキチン鎖を特異的に認識するのに対して,SHARPINのNZFはK63ポリユビキチン鎖との結合能を有する.TNFαによる細胞死誘導に対して,LUBACはE3リガーゼ活性を介して抑制的に働くが,そのためにはTNFα受容体(TNFR1)複合体へLUBACがリクルートされることが必要であり,SHARPINのNZFを介したK63ポリユビキチン鎖結合能が重要である12).Sharpinを欠損したcpdm(chronic proliferative dermatitis)マウスは,HoipおよびHoil-1l欠損マウスと異なり正常に生まれてくるが,皮膚表皮細胞(ケラチノサイト)の細胞死亢進を伴う慢性皮膚炎を発症することから,個体レベルでのLUBACの細胞死抑制機能を探索する際にきわめて有用なモデルマウスとして解析されている.
LUBACの安定性はSHARPIN, HOIL-1LおよびHOIP三者すべての発現レベルにより決定されており,アクセサリー分子であるSHARPINやHOIL-1Lの欠如は,E3リガーゼ活性を持つHOIPの量を減少させ,LUBACとしての機能を低下させる.また,SHARPINおよびHOIL-1Lとの結合はHOIPの立体構造変化を伴い,E3リガーゼ活性の自己阻害から解放し,E2-ユビキチンとの会合を容易にする.現時点では,細胞内におけるHOIPの存在量とE3リガーゼ活性を維持するためにLUBACは複合体として存在していると考えられる.
3. LUBACの直鎖状ユビキチン鎖形成メカニズム
ユビキチン内に存在する7個のリシン残基(K6, K11, K27, K29, K33, K48, K63)いずれかとC末端グリシン残基を介して,連続的にユビキチンが連結する従来のユビキチン鎖とは異なり,LUBACにより生成される直鎖状ユビキチン鎖は,N末端メチオニン残基(M1)のα-NH2基とC末端グリシン残基を介したペプチド結合によって形成される(図1)13).直鎖状ユビキチン鎖を形成する特異性はE2ではなく,E3であるLUBACが決定している.
直鎖状ユビキチン鎖の形成メカニズムに関しては,生化学的手法および結晶構造を用いて詳細に解析されている.RBR型のE3に属するLUBACは,RING(really interesting new gene)/HECT(homologous to the E6-AP carboxyl terminus)ハイブリッド型E3のユビキチン転移システムを持つ.HOIL-1LおよびSHARPINがUBA領域へ会合することで,HOIPの自己不活性化状態が解かれ,N末端側のRINGドメイン(RING1)によりE2-Ub複合体を捉えることが可能になる.次に,C末端側のRINGドメイン(RING2)内のシステイン残基へ,チオエステル結合を介してユビキチンを転移させる.その後,基質上のリシン残基のε-アミノ基へユビキチンを転移させる.以降,RING2からユビキチンをアクセプターとなるユビキチンへ受け渡す際には,RBRドメインよりさらにC末端に配置するLDD(linear ubiquitin chain determining domain)と呼ばれる領域でアクセプター側のユビキチンを捕捉し,ユビキチンN末端α-NH2基とC末端を特異的にペプチド結合させることで直鎖状ユビキチン鎖の生成を可能としている14–16).
LUBACによる直鎖状ユビキチン鎖修飾は,NF-κBシグナルの活性化に必須であることから,免疫応答や炎症システムに関連する特殊なタイプのユビキチン鎖修飾と考えられている.これまでにHOIPやHOIL-1Lに変異を有し,LUBAC機能が低下した患者は自己炎症性疾患および免疫不全症を発症することが報告されており,臨床的にも重要な遺伝性の疾患因子として認識され始めている17, 18).したがって,細胞レベルのみならず個体レベルでのLUBACの生理機能を解明していくことが必要であるであることはいうまでもない.基本的なLUBACの機能的役割として,古典的NF-κBシグナルへの活性化に加え,細胞死制御および我々が最近解明した自己炎症制御におけるTリンパ球内でのLUBACの役割を中心に解説したい.
1)古典的NF-κB経路の活性化
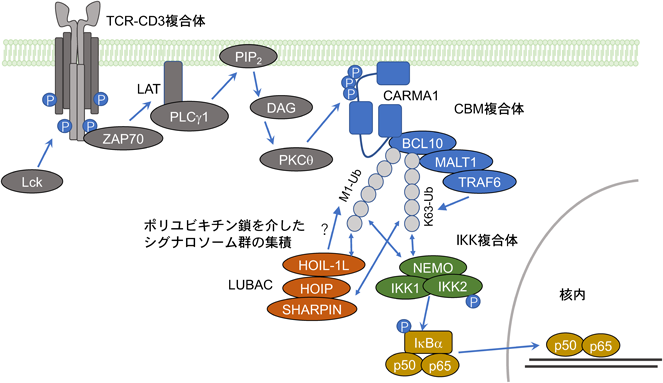
LUBACの機能に関しては,細胞の増殖や生存,その他多くの生理現象に重要な転写因子NF-κBの活性化機構への関与が最もよく知られている.NF-κBはRHD(Rel homology domain)を含む五つのタンパク質,RelA(p65),RelB, c-Rel, p105/p50(NF-κB1),p100/p52(NF-κB2)から構成され,ホモもしくはヘテロ二量体として核内へ移行し転写因子として機能する.通常,NF-κBはRHDを介して結合した阻害タンパク質IκB(inhibitor κB)によって,二量体形成および核内移行が抑制され,不活性状態として細胞質側に存在するが,細胞外からNF-κB活性化シグナルが入るとIκBはプロテアソームによって分解され,その後NF-κBが活性化される.NF-κBの活性化経路は,IKK(IκB kinase)1(IKKα),IKK2(IKKβ),NEMO(IKKγ)の三者からなるIKK複合体が介在する古典的経路と,NIK(NF-κB-inducing kinase)とIKK1が介在する非古典的経路に区別されるが,そのうちLUBACは古典的経路に関与することが知られている.古典的経路を利用する細胞外刺激はTNFα, IL-1βやLPSなどが知られており,ここでは詳細に解析されているTNFα刺激時のNF-κBシグナル伝達過程におけるLUBACの役割を例に解説する.
TNFαとその受容体(TNFR1)との結合により,TNFR1の細胞質側ではTRADD(TNFR-associated death domain)やRIPK1(receptor interacting protein 1 kinase)に加え,E3リガーゼであるTRAF2(TNF receptor-associated factor 2)やcIAP1/2(cellular inhibitor of apoptosis protein 1/2)がリクルートされることで,RIPK1上にK63やK11ユビキチン鎖が形成される.このようにしてTNFR1複合体近傍に形成されたK63ユビキチン鎖をプラットフォームにして,さらにユビキチン結合ドメインを有するシグナル因子が集積してくる(TNFR1 complex Iの形成).TAK1-TAB1-TAB2/3複合体はTAB2/3のzinc finger(NZF)ドメインを介して,IKK複合体はNEMOのC末端zinc finger(ZF)ドメインを介して,LUBAC複合体はHOIPのNZF1とSHARPINのNZFを介してTNFR1複合体近傍へリクルートされる.キナーゼであるTAK1はIKK複合体中のIKK2をリン酸化する.また,LUBACはRIPK1やIKK複合体中のNEMOを基質とし,直鎖状ユビキチン鎖を付加する.形成された直鎖状ユビキチン鎖は,さらにHOIL-1LのNZFドメインを介したLUBACのリクルートを亢進させることが予想される.NEMOに付加された直鎖状ユビキチン鎖はさらに別のIKK複合体中のNEMOのUBANドメインによって認識されることで,IKK複合体どうしが近接するようになる.その結果としてIKK2のトランス自己リン酸化(trans-autophosphorylation)が生じることで,IKK複合体が活性化すると考えられている7).
2)細胞死抑制
慢性皮膚炎を発症する突然変異マウスcpdmの原因遺伝子Sharpin(cpdmでは第1エクソンのフレームシフト変異により欠失)がコードするタンパク質は,のちにLUBACの構成因子であることが発見され,LUBACが細胞死抑制能を持つことが個体レベルで示された5).cpdmは生後3週齢ごろから過角化やケラチノサイトの細胞死亢進を伴う全身性の皮膚炎を発症する.また,肝臓,肺,食道や胃など多くの臓器で炎症症状を呈する.血中や免疫組織内での好中球や好酸球といった顆粒球の増加症が認められる一方,血中免疫グロブリン値の低下やパイエル板の欠損,T細胞の減少など獲得免疫系の機能不全を示唆する所見が認められる19, 20).LUBACが関与する古典的NF-κB経路はTNFα,リンホトキシン,TRAILなど複数のTNFスーパーファミリーに属する生理活性物質により誘導可能であり,SHARPINの欠損によりこれらの機能が減弱した結果に加え,二次的な炎症反応が所見として総合的に現れていると考えられる.皮膚炎に関していえば,皮膚特異的Sharpin欠損マウスがcpdmと同様の皮膚炎を呈することや,cpdmマウスの皮膚特異的にTNFR1を欠失させることで皮膚炎が改善することから,SHARPINの発現が,TNFα刺激から恒常的にケラチノサイトの細胞死を抑制し,皮膚組織を保護していることが示されている21, 22).
RelBやIκBα欠損マウス,Nemoヘテロ欠損マウス(X染色体上にコードされるため雌のみ),皮膚特異的Ikk2欠損マウスなどはcpdmと類似した皮膚炎を発症することから,IKK複合体活性化を介したNF-κBシグナルの亢進と抗アポトーシス因子(cIAP1, cIAP2, Bcl-2, FLIPなど)の発現亢進が,LUBACの細胞死抑制機構の理由の一つであると考えられるが,加えて細胞死の実行機構を直接的に制御している可能性がある.TNFαで刺激した際,NemoやIkk2欠損MEF細胞と比較して,LUBACの構成因子を欠損した細胞の方が明らかに高い細胞死易感受性を示す.また個体レベルにおいても,Ikk2, p65, Nemo欠損マウスと比べて,Hoil-1lやHoip欠損マウスはより早期(E10.5)に耐性致死となる事実からも,IKK複合体-NF-κBシグナル活性化経路とは異なる細胞死制御機構にLUBACが関与することは明らかである12, 23).細胞死抑制機構の詳細なメカニズムについてはまだ不明な点が多いが,RIPK1の直鎖状ユビキチン鎖修飾の重要性が示唆されている24).前述したとおり,RIPK1はTNFαがTNFR1へ結合した際にTNFR1の細胞質領域へリクルートされ,cIAP1/2やTRAF2, LUBACによってポリユビキチン化を受けるアダプター分子である.cIAP1/2やLUBACを介したユビキチン鎖形成の阻害や脱ユビキチン化酵素CYLDによるK63鎖の切断は,TNFR1 complex Iの不安定化へ導き,結果としてTNFR1 complex IからRIPK1が細胞質内へ放出される.その後,Caspase 8, FADD(Fas-associated death domain protein),RIPK3とともにcomplex IIが形成され,Caspase 8依存的なアポトーシスやRIPK3-MLKL(mixed lineage kinase domain-like protein)依存的なネクロプトーシスといったプログラム細胞死が誘導される.RIPK1の直鎖状ユビキチン鎖修飾によってTNFRI complex Iがなぜ安定化するのかについては明らかではないが,RIPK1のリン酸化がその決定に重要とされている.NEMOのUBANドメインを介してリクルートされたIKK1やIKK2がTNFRI complex I中のRIPK1をリン酸化することで複合体の安定性を高めることや,NEMOがアダプター分子であるTANKやNAP1を介して,TBK1とIKKεをTNFR1 complex I中へリクルートし,RIPK1をリン酸化することで細胞死への移行が抑制されることなどが報告されている25, 26).
3)Tリンパ球におけるLUBACの機能
LUBACおよびその構成因子の発現はすべての細胞で一様に認められるが,個体レベルでかつ臓器別に比較すると,胸腺やリンパ節,脾臓などできわめて発現が高い.すなわち,免疫組織に存在するリンパ球で高発現していることを鑑み,我々はLUBAC構成因子のコンディショナルノックアウトマウスを樹立し,Bリンパ球やTリンパ球で特異的にLUBACの機能を低下させ,その生理機能への影響を報告してきた.最近筆者らは,Tリンパ球でのLUBACが,自己免疫疾患や自己炎症性疾患など炎症性疾患の発症制御に重要であることを明らかにしており,本項ではその概要を解説する.
獲得免疫細胞であるTリンパ球は,細胞膜に発現するT細胞受容体(TCR)を介して外来抗原とMHCを認識し活性化することができる.TCR刺激時にLUBACの機能低下がもたらす影響を観察したところ,SHARPINやHOIPを欠損したTリンパ球ハイブリドーマやヒトリンパ腫細胞JurkatではNF-κB転写活性の著減に加え,刺激依存的なIL-2の産生が低下していた.興味深いことにHOIPの欠損によりNF-κB転写活性が完全に消失しているのに対して,SHARPINの欠損ではわずかに転写活性が保持されており,LUBACの安定性の低下とHOIPの残存活性を反映したものであると考えられる.また,SHARPINを欠損したマウスTリンパ球ではRelAのリン酸化やIκBαの分解がわずかに阻害されており,Tリンパ球活性化マーカーである細胞膜上CD25やCD69の発現の低下も認められた.以上の結果は,TCRシグナル下流NF-κB経路を介したTリンパ球の活性化においてLUBAC複合体の存在が必須であることを示している22).
抗原認識後,細胞膜上のTCR-CD3複合体内では細胞質領域CD3-ITAMモチーフに含まれるチロシンがSrcファミリーチロシンキナーゼLckによってリン酸化され,リン酸化チロシンに対してSykファミリーチロシンキナーゼZAP70がN末端領域のSH2ドメインを介して会合し,活性化することでLATなどさらなる下流のアダプター分子がリン酸化を受ける.TCR下流のIKK複合体依存的なNF-κB経路は,またさらに細胞膜近傍へとリクルートされるPKCθとその下流に存在する二つのシグナロソーム,CBM(CARMA1-BCL10-MALT1)複合体とLUBACによって誘導される.PKCθがCARMA1をリン酸化しCBM複合体の形成と細胞膜近傍への集積を促す.CBM複合体中のBCL10がK63ユビキチン化を受けていることや,MALT1がTRAF6との結合能を有することなどから,CBM複合体が自身に結合したユビキチン鎖を介して(TNFR1複合体でいえばRIPK1のような役割),LUBACおよびIKK複合体をリクルートし,活性化に導いているのかもしれない(図2).また,CBM複合体中のMALT1はパラカスパーゼ活性を有しており,LUBACの構成因子であるHOIL-1Lを切断し,LUBACの活性に影響を与えていることが報告されている27).しかしながら,筆者らはJurkatを用いた解析から,TCR刺激によるNF-κB経路の活性化にはユビキチン鎖結合能は必要であるが,E3酵素活性は必要ない,つまりE3リガーゼ非依存的なLUBACの役割が存在する可能性を示唆する結果も得ており,当該シグナル経路でLUBACが必要とされる理由についてはいまだ明らかにされておらず,さらなる解析が期待される22, 28).
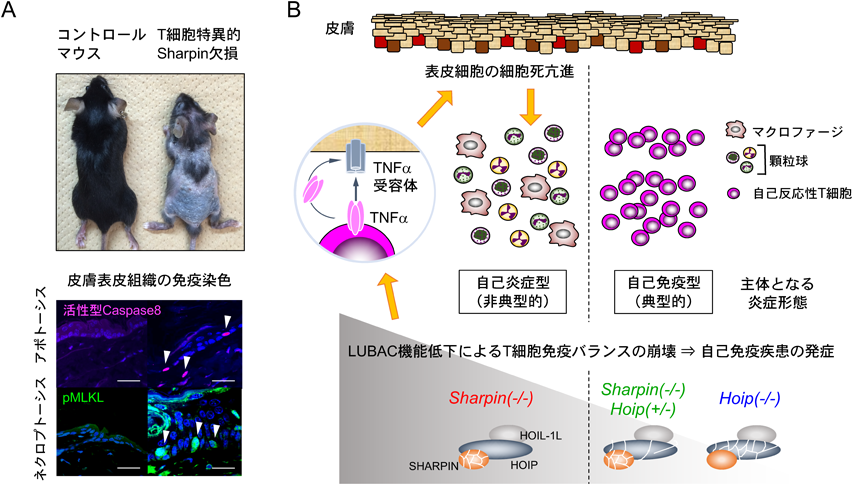
前述したとおり,Sharpin欠損マウスcpdmはケラチノサイトの細胞死が亢進することで全身性の皮膚炎を発症するが,近年複数のグループから,cpdmでは制御性T細胞(Treg;炎症を抑制するタイプのTリンパ球)の細胞数が減少しており,Tリンパ球依存性の自己免疫疾患を発症している可能性が示唆されていた29–31).筆者らはcpdmの病態形成におけるTリンパ球の影響を明確にするため,Sharpin遺伝子のコンディショナルノックアウトマウスおよびコンディショナルトランスジェニックマウスを用いて遺伝学的に詳細な解析を行った.その結果,SHARPINをTリンパ球で欠失させるとcpdmと同様の皮膚炎を発症することや,cpdmのTリンパ球で特異的にSHARPINを発現させると皮膚炎が改善することなどを明らかにし,cpdmの皮膚炎の増悪化にTリンパ球が影響していることを明らかにしている(図3A).さらにTreg特異的Sharpin欠損マウスも加えた解析から,Tリンパ球内でのSHARPINの消失は,Tregの分化,増殖,炎症抑制能の重度の機能低下をもたらし,それに伴い活性化するエフェクターTリンパ球により炎症が誘発されている(T細胞免疫バランスの崩壊)ことを見いだしている22).
本研究には炎症性疾患の病因と病態形成に関連した重要な伏線がある.Tリンパ球やBリンパ球(獲得免疫系細胞)が発症に関与する自己免疫疾患とは対照的に,自己炎症性疾患は,自然免疫系細胞の活性化と炎症性サイトカインが発症の原因となる遺伝性の炎症性疾患である.従来cpdmの病巣部ではリンパ球の関与が認められないため,細胞死を起因とする自己炎症性の機序で皮膚炎が発症すると考えられていた.しかしながら,筆者らはTリンパ球やTreg特異的Sharpin欠損マウスが示す病態形成の詳細な解析から,末梢組織に浸潤した活性化Tリンパ球が炎症性サイトカインTNFαを介してケラチノサイトの細胞死を誘導し,自然免疫系細胞の浸潤を亢進させることで,見かけ上,自己炎症性疾患(cpdm様)の所見を示すことにつながる新たな炎症病態メカニズムを提唱している(図3A).筆者らはT細胞免疫バランスをさらに悪化させ,より重篤な自己免疫疾患をマウスで発症させる目的で,Treg特異的Hoip欠損マウスを用いて,新たに2系統のマウスSharpin(−/−)Hoip(+/−),Hoip(−/−)を作製した(図3B).これはLUBACがT細胞内で複合体として機能しており,その機能がHOIPの量に依存していることを利用している.Sharpin(−/−)Hoip(+/−)はSharpin(−/−)よりも早期に皮膚炎を発症し,Hoip(−/−)はTregが存在しないFoxp3欠損マウス(Scurfyマウス)と同様の表現型を示し,3週齢前後で致死となる.これらのマウスの皮膚では,Sharpin遺伝子単独欠損時とは異なり,Tリンパ球主体の典型的な自己免疫性の病理像を示した22).したがって,筆者らが見いだした自己炎症様の炎症所見は,SHARPINの欠損による緩和なLUBAC機能低下(HOIPの残存活性あり)がTリンパ球で起きた場合に主体的に観察される病理像であると考えられる(図3B).これらの結果はcpdmが自己免疫疾患と自己炎症性疾患を同時に発症している炎症モデルであることに加え,T細胞の機能異常が自己炎症性皮膚炎を引き起こすことを初めて明らかとしている.これまで報告されているHOIPやHOIL-1Lの遺伝子変異からLUBACの発現が低下した患者では,自己炎症性疾患は発症するが,皮膚炎を主体とした所見は報告されていない.種間における細胞の感受性の違いによるものと考えられるが,今回明らかにされたT細胞による自己炎症の発症メカニズムがヒト炎症性疾患で起きている可能性を探索する必要があろう.炎症を抑え,生体の恒常性を維持するマスター因子として,個体レベルでのLUBACの生理学的存在意義が明確になってきたと思われる.
引用文献References
1) Hershko, A. & Ciechanover, A. (1998) The ubiquitin system. Annu. Rev. Biochem., 67, 425–479.
2) Komander, D. & Rape, M. (2012) The ubiquitin code. Annu. Rev. Biochem., 81, 203–229.
3) Sasaki, K. & Iwai, K. (2015) Roles of linear ubiquitinylation, a crucial regulator of NF-κB and cell death, in the immune system. Immunol. Rev., 266, 175–189.
4) Tokunaga, F., Sakata, S., Saeki, Y., Satomi, Y., Kirisako, T., Kamei, K., Nakagawa, T., Kato, M., Murata, S., Yamaoka, S., et al. (2009) Involvement of linear polyubiquitylation of NEMO in NF-κB activation. Nat. Cell Biol., 11, 123–132.
5) Tokunaga, F., Nakagawa, T., Nakahara, M., Saeki, Y., Taniguchi, M., Sakata, S., Tanaka, K., Nakano, H., & Iwai, K. (2011) SHARPIN is a component of the NF-κB-activating linear ubiquitin chain assembly complex. Nature, 471, 633–636.
6) Fujita, H., Tokunaga, A., Shimizu, S., Whiting, A.L., Aguilar-Alonso, F., Takagi, K., Walinda, E., Sasaki, Y., Shimokawa, T., Mizushima, T., et al. (2018) Cooperative domain formation by homologous motifs in HOIL-1L and SHARPIN plays a crucial role in LUBAC stabilization. Cell Rep., 23, 1192–1204.
7) Fujita, H., Rahighi, S., Akita, M., Kato, R., Sasaki, Y., Wakatsuki, S., & Iwai, K. (2014) Mechanism underlying IκB kinase activation mediated by the linear ubiquitin chain assembly complex. Mol. Cell. Biol., 34, 1322–1335.
8) Elliott, P.R., Leske, D., Hrdinka, M., Bagola, K., Fiil, B.K., McLaughlin, S.H., Wagstaff, J., Volkmar, N., Christianson, J.C., Kessler, B.M., et al. (2016) SPATA2 Links CYLD to LUBAC, Activates CYLD, and Controls LUBAC Signaling. Mol. Cell, 63, 990–1005.
9) Takiuchi, T., Nakagawa, T., Tamiya, H., Fujita, H., Sasaki, Y., Saeki, Y., Takeda, H., Sawasaki, T., Buchberger, A., Kimura, T., et al. (2014) Suppression of LUBAC-mediated linear ubiquitination by a specific interaction between LUBAC and the deubiquitinases CYLD and OTULIN. Genes Cells, 19, 254–272.
10) Kelsall, I.R., Zhang, J., Knebel, A., Arthur, J.S.C., & Cohen, P. (2019) The E3 ligase HOIL-1 catalyses ester bond formation between ubiquitin and components of the Myddosome in mammalian cells. Proc. Natl. Acad. Sci. USA, 116, 13293–13298.
11) Sato, Y., Fujita, H., Yoshikawa, A., Yamashita, M., Yamagata, A., Kaiser, S.E., Iwai, K., & Fukai, S. (2011) Specific recognition of linear ubiquitin chains by the Npl4 zinc finger (NZF) domain of the HOIL-1L subunit of the linear ubiquitin chain assembly complex. Proc. Natl. Acad. Sci. USA, 108, 20520–20525.
12) Shimizu, S., Fujita, H., Sasaki, Y., Tsuruyama, T., Fukuda, K., & Iwai, K. (2016) Differential involvement of the Npl4 zinc finger domains of SHARPIN and HOIL-1L in linear ubiquitin chain assembly complex-mediated cell death protection. Mol. Cell. Biol., 36, 1569–1583.
13) Kirisako, T., Kamei, K., Murata, S., Kato, M., Fukumoto, H., Kanie, M., Sano, S., Tokunaga, F., Tanaka, K., & Iwai, K. (2006) A ubiquitin ligase complex assembles linear polyubiquitin chains. EMBO J., 25, 4877–4887.
14) Lechtenberg, B.C., Rajput, A., Sanishvili, R., Dobaczewska, M.K., Ware, C.F., Mace, P.D., & Riedl, S.J. (2016) Structure of a HOIP/E2~ubiquitin complex reveals RBR E3 ligase mechanism and regulation. Nature, 529, 546–550.
15) Smit, J.J., Monteferrario, D., Noordermeer, S.M., van Dijk, W.J., van der Reijden, B.A., & Sixma, T.K. (2012) The E3 ligase HOIP specifies linear ubiquitin chain assembly through its RING-IBR-RING domain and the unique LDD extension. EMBO J., 31, 3833–3844.
16) Stieglitz, B., Rana, R.R., Koliopoulos, M.G., Morris-Davies, A.C., Schaeffer, V., Christodoulou, E., Howell, S., Brown, N.R., Dikic, I., & Rittinger, K. (2013) Structural basis for ligase-specific conjugation of linear ubiquitin chains by HOIP. Nature, 503, 422–426.
17) Boisson, B., Laplantine, E., Dobbs, K., Cobat, A., Tarantino, N., Hazen, M., Lidov, H.G., Hopkins, G., Du, L., Belkadi, A., et al. (2015) Human HOIP and LUBAC deficiency underlies autoinflammation, immunodeficiency, amylopectinosis, and lymphangiectasia. J. Exp. Med., 212, 939–951.
18) Boisson, B., Laplantine, E., Prando, C., Giliani, S., Israelsson, E., Xu, Z., Abhyankar, A., Israël, L., Trevejo-Nunez, G., Bogunovic, D., et al. (2012) Immunodeficiency, autoinflammation and amylopectinosis in humans with inherited HOIL-1 and LUBAC deficiency. Nat. Immunol., 13, 1178–1186.
19) Gijbels, M.J., Zurcher, C., Kraal, G., Elliott, G.R., HogenEsch, H., Schijff, G., Savelkoul, H.F., & Bruijnzeel, P.L. (1996) Pathogenesis of skin lesions in mice with chronic proliferative dermatitis (cpdm/cpdm). Am. J. Pathol., 148, 941–950.
20) HogenEsch, H., Gijbels, M.J., Offerman, E., van Hooft, J., van Bekkum, D.W., & Zurcher, C. (1993) A spontaneous mutation characterized by chronic proliferative dermatitis in C57BL mice. Am. J. Pathol., 143, 972–982.
21) Rickard, J.A., Anderton, H., Etemadi, N., Nachbur, U., Darding, M., Peltzer, N., Lalaoui, N., Lawlor, K.E., Vanyai, H., Hall, C., et al. (2014) TNFR1-dependent cell death drives inflammation in Sharpin-deficient mice. eLife, 3, 3.
22) Sasaki, K., Himeno, A., Nakagawa, T., Sasaki, Y., Kiyonari, H., & Iwai, K. (2019) Modulation of autoimmune pathogenesis by T cell-triggered inflammatory cell death. Nat. Commun., 10, 3878.
23) Peltzer, N., Rieser, E., Taraborrelli, L., Draber, P., Darding, M., Pernaute, B., Shimizu, Y., Sarr, A., Draberova, H., Montinaro, A., et al. (2014) HOIP deficiency causes embryonic lethality by aberrant TNFR1-mediated endothelial cell death. Cell Rep., 9, 153–165.
24) Witt, A. & Vucic, D. (2017) Diverse ubiquitin linkages regulate RIP kinases-mediated inflammatory and cell death signaling. Cell Death Differ., 24, 1160–1171.
25) Dondelinger, Y., Jouan-Lanhouet, S., Divert, T., Theatre, E., Bertin, J., Gough, P.J., Giansanti, P., Heck, A.J., Dejardin, E., Vandenabeele, P., et al. (2015) NF-κB-independent role of IKKα/IKKβ in preventing RIPK1 kinase-dependent apoptotic and necroptotic cell death during TNF signaling. Mol. Cell, 60, 63–76.
26) Lafont, E., Draber, P., Rieser, E., Reichert, M., Kupka, S., de Miguel, D., Draberova, H., von Mässenhausen, A., Bhamra, A., Henderson, S., et al. (2018) TBK1 and IKKε prevent TNF-induced cell death by RIPK1 phosphorylation. Nat. Cell Biol., 20, 1389–1399.
27) Klein, T., Fung, S.Y., Renner, F., Blank, M.A., Dufour, A., Kang, S., Bolger-Munro, M., Scurll, J.M., Priatel, J.J., Schweigler, P., et al. (2015) The paracaspase MALT1 cleaves HOIL1 reducing linear ubiquitination by LUBAC to dampen lymphocyte NF-κB signalling. Nat. Commun., 6, 8777.
28) Beyaert, R. (2014) An E3 ubiquitin ligase-independent role of LUBAC. Blood, 123, 2131–2133.
29) Park, Y., Jin, H.S., Lopez, J., Lee, J., Liao, L., Elly, C., & Liu, Y.C. (2016) SHARPIN controls regulatory T cells by negatively modulating the T cell antigen receptor complex. Nat. Immunol., 17, 286–296.
30) Redecke, V., Chaturvedi, V., Kuriakose, J., & Hacker, H. (2016) SHARPIN controls the development of regulatory T cells. Immunology, 148, 216–226.
31) Teh, C.E., Lalaoui, N., Jain, R., Policheni, A.N., Heinlein, M., Alvarez-Diaz, S., Sheridan, J.M., Rieser, E., Deuser, S., Darding, M., et al. (2016) Linear ubiquitin chain assembly complex coordinates late thymic T-cell differentiation and regulatory T-cell homeostasis. Nat. Commun., 7, 13353.
32) Noad, J., von der Malsburg, A., Pathe, C., Michel, M.A., Komander, D., & Randow, F. (2017) LUBAC-synthesized linear ubiquitin chains restrict cytosol-invading bacteria by activating autophagy and NF-κB. Nat. Microbiol., 2, 17063.
33) Rodgers, M.A., Bowman, J.W., Fujita, H., Orazio, N., Shi, M., Liang, Q., Amatya, R., Kelly, T.J., Iwai, K., Ting, J., et al. (2014) The linear ubiquitin assembly complex (LUBAC) is essential for NLRP3 inflammasome activation. J. Exp. Med., 211, 1333–1347.
34) Heger, K., Wickliffe, K.E., Ndoja, A., Zhang, J., Murthy, A., Dugger, D.L., Maltzman, A., de Sousa E Melo, F., Hung, J., Zeng, Y., et al. (2018) OTULIN limits cell death and inflammation by deubiquitinating LUBAC. Nature, 559, 120–124.