ミトコンドリアはほとんどの真核生物の細胞に含まれる細胞小器官で,酸化的リン酸化によるATPの産生の他,細胞周期,アポトーシスの制御といった多様な役割を担っている.一方でミトコンドリアは,ATP産生に伴う反応により細胞内で最大の活性酸素種(ROS)産生器官としても振る舞い,自身が産生したROSによる酸化ストレスに絶えず曝露されて損傷を受ける1).抗酸化酵素やDNA・タンパク質を修復・分解する酵素によってミトコンドリアの品質は維持されるが,品質維持能力を上回る損傷を受けた不良ミトコンドリアが細胞内に蓄積すると,細胞のアポトーシスを誘導し,パーキンソン病などの神経変性疾患を引き起こす.このような細胞損傷を回避するために,細胞はミトコンドリア選択的なオートファジー(マイトファジー)により不良ミトコンドリアを分解除去している.不良ミトコンドリアに関わるマイトファジーの制御は,若年性パーキンソン病の原因遺伝子であるPARK2とPINK1/PARK6がそれぞれコードするユビキチンリガーゼparkinとユビキチンキナーゼPTEN-induced putative kinase 1(PINK1)2, 3),そして脱ユビキチン化酵素ubiquitin-specific protease 30(USP30)が関わっている4).この制御機構は,ユビキチンのリン酸化や,非定型型ユビキチン鎖であるK6鎖が関わる複雑なものであったが,近年の機能・構造解析の成果により,多くの部分が解明されつつある.本稿では,「K6鎖特異的な脱ユビキチン化酵素USP30」,「ユビキチンやparkinをリン酸化するキナーゼPINK1」,「リン酸化ユビキチンの結合と自身のリン酸化によって活性化されるユビキチンリガーゼparkin」に関して,最新の知見を我々の研究成果を交えながら紹介したい.
2. ユビキチン鎖により制御されるミトコンドリア分解
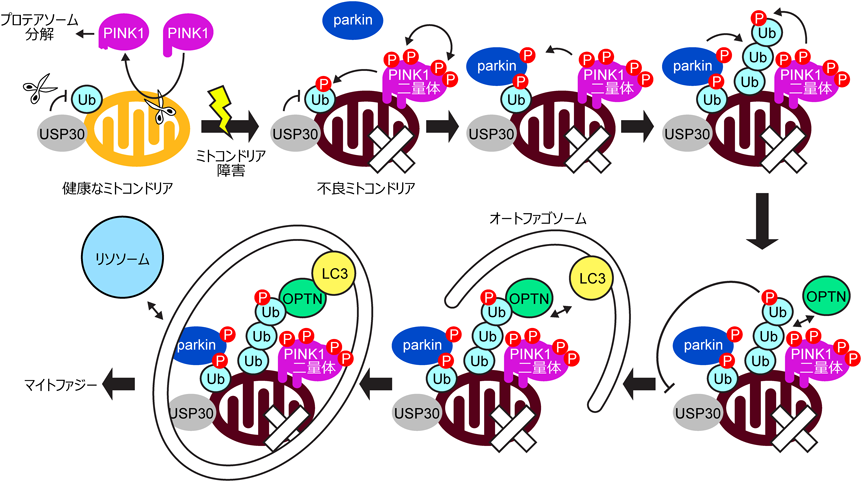
不良ミトコンドリアは,ミトコンドリア選択的なオートファジーであるマイトファジーによって分解される.一般的なオートファジーでは,細胞質に存在するタンパク質やオルガネラが非選択的にオートファゴソームに取り込まれ,オートファゴソームとリソソームが融合することでその内容物を分解する.マイトファジーはオートファジーの一種であるが,オートファゴソームがミトコンドリアを選択的に取り込むという点が特徴である.マイトファジーがミトコンドリアを選択的に取り込む過程には,ユビキチン介在経路と受容体介在経路の2種類が存在するが,不良ミトコンドリアの分解はユビキチンキナーゼPINK1とユビキチンリガーゼparkinによって活性化されるユビキチン介在性マイトファジーが担う.
PINK1はそのN末端側にミトコンドリア移行シグナルを,C末端側にキナーゼドメインを持つ,セリン/トレオニンキナーゼである.膜電位が正常に保たれた健康的なミトコンドリアでは,PINK1のN末端がミトコンドリア外膜だけでなく,ミトコンドリア内膜まで通過し,ミトコンドリア内膜のプロテアーゼによりミトコンドリア移行シグナルの切断を受ける(図1)5).移行シグナルが失われたPINK1は細胞質へと戻され,プロテアソームによる分解を受ける.一方,膜電位が低下している不良ミトコンドリアでは,PINK1のN末端は内膜を通過しないためプロテアーゼによる切断を受けず,PINK1は不良ミトコンドリアの外膜上に蓄積する.蓄積したPINK1は外膜上で二量体になりトランス自己リン酸化により活性化され6, 7),ミトコンドリア外膜上ユビキチンのSer65をリン酸化する8).parkinはリン酸化を受けたユビキチンと結合することで不良ミトコンドリアに移動し,PINK1によってリン酸化される9).parkinは通常,自己阻害型であるが,上記の過程により活性型に変換され,ミトコンドリア外膜タンパク質上にK6, K11, K48, K63鎖を付加する10).この4種類のユビキチン鎖のマイトファジーにおける役割の違いは解明されてないが,p62, optineurin(OPTN),nuclear dot protein 52 kDa(NDP52)などのオートファジーアダプタータンパク質がユビキチン鎖と結合する.これらアダプタータンパク質はオートファゴソーム上のLC3と結合する領域を持ち,オートファゴソームと不良ミトコンドリアを結びつけるため,不良ミトコンドリアはオートファゴソームに取り込まれる.続いて,オートファゴソームがリソソームと融合することにより,その内容物を分解する.
以上のように,ユビキチン鎖はマイトファジーを亢進するが,逆に脱ユビキチン化酵素によるユビキチン鎖の切断はマイトファジーを遅延する.USPファミリーに属する脱ユビキチン化酵素USP30はN末端にミトコンドリア移行シグナルを,C末端側に脱ユビキチン化活性ドメインを持ち,現在知られている中ではミトコンドリア外膜上に常に存在する唯一の脱ユビキチン化酵素である4).USP30はparkinと拮抗的に働き,健康なミトコンドリア外膜上のユビキチン鎖の量が増えすぎないように,一定に保っていると考えられている11).USP30を過剰発現させた細胞では,parkinによって合成されるユビキチン鎖のうち特にK6鎖が減少し,USP30を欠失させた細胞では,ミトコンドリア外膜上に主にK6鎖が増加する.さらに,精製したUSP30はK6, K11, K48, K63鎖を切断するが,特にK6鎖に対して強い活性を持つ10, 12).一方,USP30はリン酸化を受けたK6鎖に対する活性が減弱する12).PINK1はparkinによって付加されたユビキチン鎖もリン酸化するが,このリン酸化はparkinの活性化だけでなく,USP30の活性を阻害することでparkinにより伸長されたユビキチン鎖を保護するという役割も持つと考えられている.
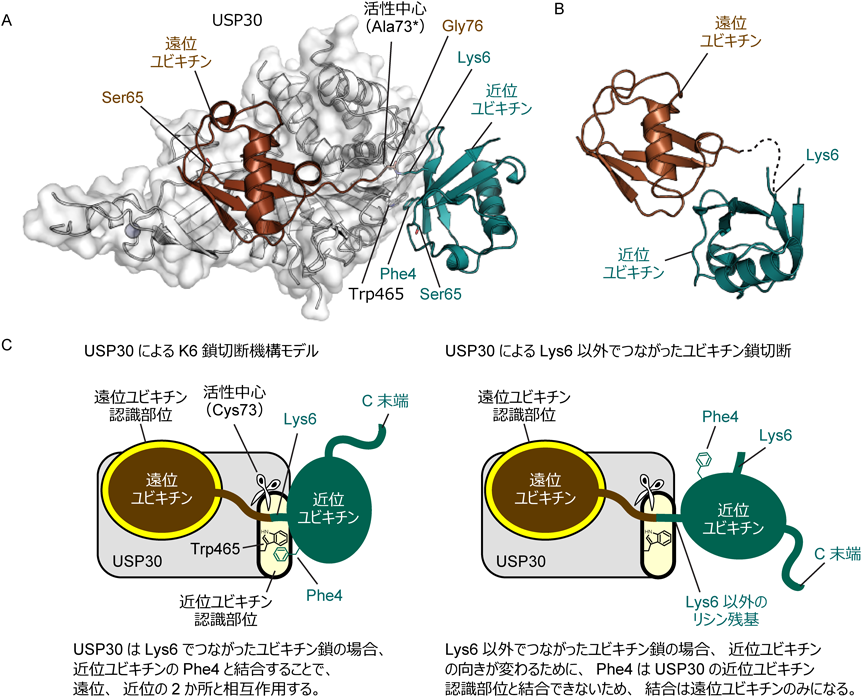
著者らは,ヒト由来よりも,ゼブラフィッシュ由来のUSP30(zUSP30)の熱安定性が高く発現量も多いことから,zUSP30を用いて生化学的な実験を行った13).ユビキチンが2個つながったユビキチン鎖(Ub2)を用いてzUSP30のK6鎖,K11鎖に対する切断活性を測定したところ,zUSP30のK6-Ub2に対する切断活性(kcat/Km)はK11-Ub2に対する活性よりも8.6倍強いことがわかった.また,zUSP30の活性中心であるシステイン残基Cys73をアラニン残基(C73A)に置換して不活化した上で,K6-, K11-, K48-Ub2との解離定数(Kd)を測定したところ,zUSP30はK6-Ub2のみと強く結合する(Kd=2.7 μM)ことがわかった.プロテアーゼが基質を切断するためには,少なくとも結合,切断,解離のステップが存在するが,不活性型zUSP30がK6-Ub2のみと強く結合することから,zUSP30は結合の段階でK6鎖を特異的に見分けていると考えられる.
続いてUSP30によるK6鎖特異的切断機構を明らかにするため,zUSP30(C73A)とK6-Ub2との複合体(zUSP30–K6-Ub2)の結晶構造解析を行った(図2A)13).K6-Ub2の二つのユビキチンのうち,ユビキチン鎖が基質を修飾する際に基質から遠い方を遠位ユビキチン,近い方を近位ユビキチンと呼ぶが,K6-Ub2単体では遠位ユビキチンと近位ユビキチンが疎水性相互作用により結合した,コンパクトな構造をとることが知られていた(図2B)14).しかし,zUSP30–K6-Ub2の結晶構造では,zUSP30は遠位と近位のユビキチンを同時に結合し,K6-Ub2は長く引き伸ばされていた.つまり,zUSP30はK6鎖を引き伸ばした状態で固定し,K6鎖を切断すると考えられる.
zUSP30によるユビキチンの認識を詳しくみると,遠位ユビキチンの認識はこれまでに構造解析された他のUSPファミリーによるユビキチンの認識とよく似た様式であった.また,遠位ユビキチンの認識に関わる残基に変異を導入すると,K6, K11鎖に対する活性が同程度減少した.このため,遠位ユビキチンの認識は,K6鎖特異的切断ではなく,zUSP30によるユビキチン鎖全般への活性に関わるということがわかった.一方,近位ユビキチンの認識は,これまでに報告されたUSPファミリーによるユビキチン認識機構とは大きく異なるものであった.近位ユビキチンの認識に特に重要なのは,他のUSPファミリーでは別のアミノ酸に置換されている一方,USP30では種を超えて保存されているzUSP30のTrp465(ヒトではTrp475)による近位ユビキチンのPhe4の認識であった.このTrp465をアラニンに置換するとK6-Ub2に対する切断活性が大幅に減少するが,K11-Ub2に対する切断活性はあまり変わらないため,zUSP30はK6-Ub2とK11-Ub2に対する活性がほぼ等しくなり特異性を失った.つまり,zUSP30はK6-Ub2の遠位と近位ユビキチンの二つと同時に結合することで,K6鎖と強く結合し切断するが,K6鎖以外のユビキチン鎖では近位ユビキチンの配向が変わるために結合は失われ,遠位ユビキチンのみとの結合でユビキチン鎖を切断するため弱い活性となる,というメカニズムが明らかとなった(図2C).
続いて,PINK1によるユビキチンのSer65のリン酸化が,USP30の活性を減弱するメカニズムについて解説する.結晶構造から,USP30は遠位ユビキチンのSer65と相互作用する一方,近位ユビキチンのSer65とは相互作用しないことがわかった(図2A).片方のユビキチンのみをリン酸化して,USP30によるK6-Ub2に対する活性を確認したところ,遠位ユビキチンのリン酸化は活性を減弱するが,近位ユビキチンのリン酸化は活性に影響を与えないことがわかった.つまり,遠位ユビキチンのリン酸化により,本来の遠位ユビキチンとの相互作用が弱まりUSP30の活性は減弱する.PINK1は不良ミトコンドリア上において,遠位のユビキチンを優先的にリン酸化するため,PINK1によるリン酸化はUSP30の活性を効率的に阻害できると考えられる.
なお,著者らの論文と同時に,Komanderらの研究グループから,ヒトUSP30とK6-Ub2との複合体の結晶構造に関する論文が出版された15).彼らは,活性に直接関与しない挿入領域を切り詰めることでヒトUSP30を安定化し,K6-Ub2との複合体の結晶を得ているが,彼らの結果と著者らの結果はほぼ一致しており,ヒトとゼブラフィッシュでUSP30によるK6鎖切断機構や,ユビキチンのリン酸化によるUSP30の活性阻害機構は同様であることが証明された.また,USP30はマイトファジーのシグナルであるユビキチン鎖を切断するため,その阻害剤はマイトファジーを亢進し,パーキンソン病に対する治療薬の候補として注目されている.したがってUSP30の構造情報は,パーキンソン病の治療薬開発につながることが期待される11, 13, 15).
4. PINK1の活性化およびユビキチンのリン酸化機構
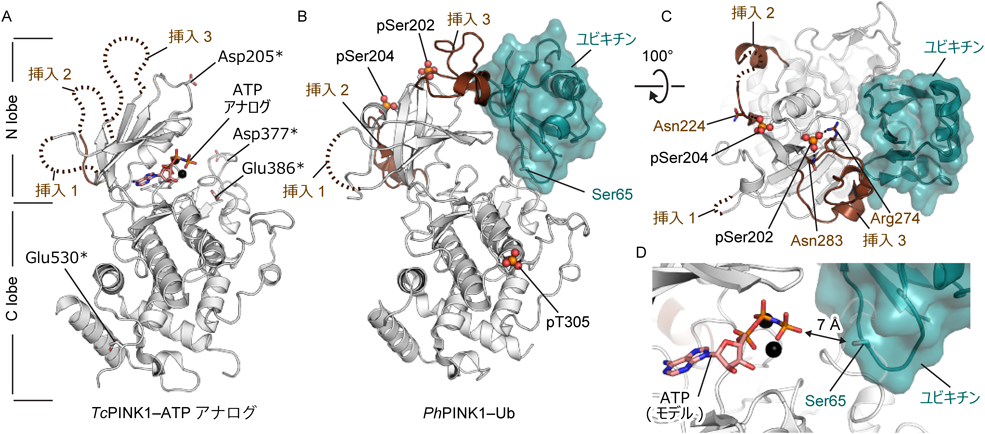
PINK1はヒトから線虫まで保存されており,そのドメイン構成もよく似ているが,C末端側のキナーゼドメインを切り出して発現系を構築すると,昆虫由来のPINK1のみ高い可溶性と強いリン酸化活性を有する.そこで著者らのグループはコクヌストモドキ(Tribolium castaneum)由来のPINK1(TcPINK1)を用いて,機能・構造解析を行った.結晶構造解析には均一性の高いサンプルが要求されるが,PINK1を大腸菌で発現させると細胞内で複数か所のセリン/トレオニン残基が自己リン酸化されて不均一なリン酸化状態となり,結晶が得られなかった.そこで著者らは,リン酸化される頻度の高いセリンやトレオニンを質量分析により同定し,リン酸化を模倣するような酸性残基に置換した(S205D, S377D, T386E, T530E).さらに,わずかに残ったリン酸化部位をγホスファターゼで脱リン酸化することで均一なサンプルの調製に成功した.ユビキチンのリン酸化にはPINK1の自己リン酸化が必要であるが,アスパラギン酸やグルタミン酸への置換ではリン酸化を完全に模倣することはできなかったためTcPINK1のユビキチンに対するリン酸化活性は弱い状態であったが,ATPアナログ(AMP-PNP)との複合体として結晶化を行ったところ結晶構造の決定に成功した(図3A)16).TcPINK1は一般的なキナーゼと同様に,N末端側のN lobeとC末端側のC lobeにより,ATPをはさみ込むように結合していた.一方,PINK1は一般的なキナーゼドメインに加えて,N lobeに三つの挿入領域,挿入1, 2, 3を持つが,これらはすべてディスオーダーしており,モデルを構築することができなかった.なお,著者らの結晶構造より先行してTcPINK1単体17)の結晶構造が報告されたが,著者らの方法と同様にリン酸化部位を酸性残基に置換してあり,挿入領域は大部分がディスオーダーしている点など,著者らの結果とよく似た構造であった.
著者らのグループはTcPINK1とユビキチンとの複合体の結晶化も試みたが,両者の結合が弱く,結晶を得ることはできなかった.一方,PINK1の構造解析は世界的に競争があり,Komanderらのグループからコロモジラミ(Pediculus humanus corporis)由来のPINK1(PhPINK1)とユビキチンとの複合体18)の結晶構造が報告された(図3B).キナーゼと基質の結合は一般に一過的なものであり解離しやすいため,PhPINK1-ユビキチン複合体の結晶構造解析では安定な複合体を形成するような手法が用いられていた.簡潔に述べると,1)PINK1との結合が強くなる変異をユビキチンに導入,2)PINK1-ユビキチンの結合を増強するナノボディを作成しPINK1-ユビキチン-ナノボディの複合体として結晶化,3)PINK1-ユビキチン-ナノボディに対してγホスファターゼ処理をすることでユビキチンのリン酸化に必要な自己リン酸化は残し,不必要な自己リン酸化のみを除去,という三つである.PhPINK1-ユビキチンの結晶構造では,ディスオーダーしていた挿入領域のうち挿入2, 3の大部分が構造をとり,N lobeの挿入領域以外の一部の領域も挿入2, 3との相互作用により構造変化を起こしていた(図3B).この構造変化は,PINK1のリン酸化されたSer202, Ser204(pSer202, pSer204)がそれぞれ挿入3のArg274, Asn283,挿入2のAsn224との間に水素結合を形成することで引き起こされる(図3C).挿入3がユビキチンとの相互作用に直接関わっている他,N lobeの構造変化した領域がユビキチンとの結合に関わるため,PhPINK1がSer202とSer204の自己リン酸化を受けると,ユビキチンへの親和性が高まると考えられる.この活性化機構はヒトPINK1のSer228(PhPINK1のSer202)のリン酸化がユビキチンのリン酸化に必要であるという過去の知見6)ともよく一致している.
PhPINK1-ユビキチン複合体の結晶構造中に核酸は含まれていなかったが,TcPINK1-ATPアナログ複合体と重ね合わせることでATPのモデルを置いてみると,ATPのγ位のリン酸がユビキチンSer65と7 Åの距離にあることがわかった(図3D).この距離は直接Ser65をリン酸化するには遠すぎるが,Ser65はユビキチンの柔軟なループ上の残基であるため,ATP存在下でSer65がわずかに動くことで反応が進むと考えられる.
以上の結果をまとめると,PINK1はN lobeの挿入領域がディスオーダーしているが(図3A),Ser202とSer204がリン酸化されることで挿入2, 3が構造をとるとともにN lobeの一部構造が変化し,ユビキチン結合サイトが形成される(図3B, C).ユビキチンがPINK1に結合すると,ユビキチンのSer65はATPの近傍に接近し,ATPのγ位のリン酸を受け取る(図3D),という反応機構が考えられる.なお,PINK1はparkinのubiquitin like(UBL)ドメインのSer65もリン酸化するが8),parkinのUBLドメインはその名のとおりユビキチンとよく似た構造を持ち,リン酸化を受けるSer65の位置も等しいことから,ユビキチンと同じメカニズムでリン酸化を受けると考えられる.
PINK1によるリン酸化機構についてはこれまでの研究で多くの疑問点が解明されたが,PINK1がミトコンドリア外膜上でどのように存在しているのか,という点は未解明である.PINK1はTOM複合体を通してN末端が外膜を通過し,二量体を形成して外膜上にとどまると考えられているが,その詳しい機構は明らかになっていない.TOM複合体とPINK1の複合体の構造解析により,この謎が解決されることが期待される.
著者らのグループは取り組んでいなかったが,parkinの立体構造解明の研究も世界的に非常に激しい競争があった.紙面の関係でそのすべてを紹介することはできないが,自己阻害型,リン酸化ユビキチンと結合した状態,そしてUBLがリン酸化されて活性化した状態のparkinの立体構造が,複数の研究グループから報告された.ここでは,ヒトparkinを使って行われた立体構造の成果を中心に,機能・構造解析によって得られたparkin活性化の機構について紹介したい.
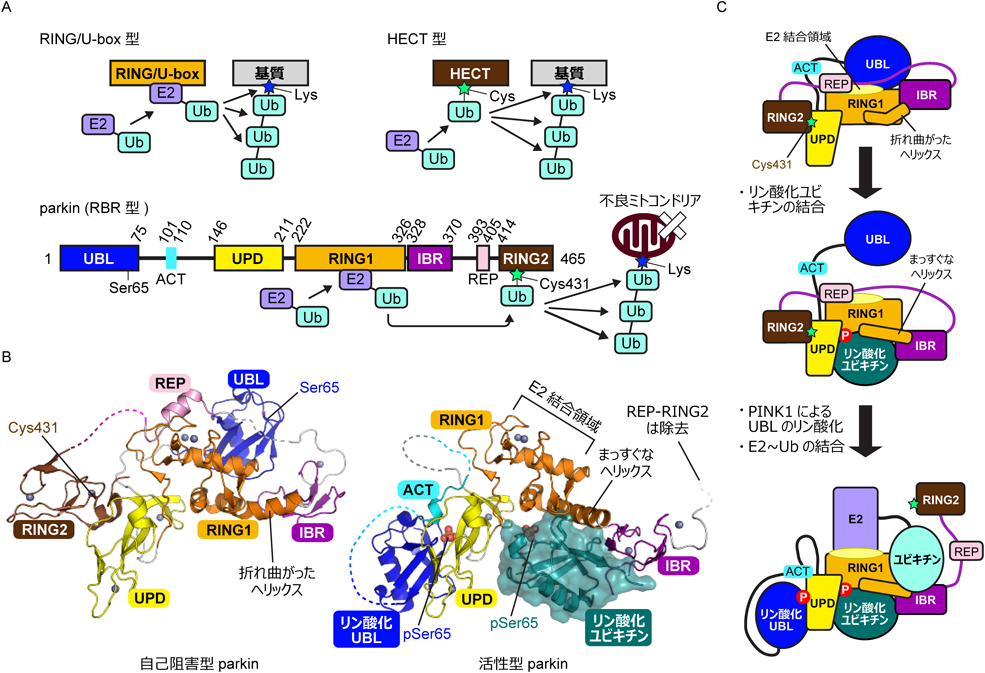
真核生物のユビキチンリガーゼは,RING型,U-box型,HECT型,RBR(RING-between-RING)型の四つに大別される(図4A).RING型とU-box型は構造と作用機序がよく似ており,ユビキチンとつながったE2(E2~Ub)と結合し,ユビキチンを基質のリシン残基へと直接受け渡す.一方HECT型は,HECTドメインの活性中心のシステイン残基がE2~Ubからユビキチンをチオエステル結合の形で受け取り,この中間体を介して基質をユビキチン化する.parkinはRBR型のユビキチンリガーゼであり,N末端側から,UBLドメイン,active element(ACT),unique parkinドメイン(UPD:RING0とも呼ばれる),RING1ドメイン,in-between-RING(IBR)ドメイン,repressor element of parkin(REP)ドメイン,RING2ドメインで構成されている(図4A).RING1ドメインはRING型E3とよく似た構造と機能を持つ.一方,RING2ドメインはRING型E3と相同性がなく,ユビキチンを受け取る活性中心のシステイン(Cys431)を持つため,機能的にはHECT型E3と似ている.その反応機構は,1)E2~UbがparkinのRING1ドメインと結合する,2)ユビキチンがRING2ドメインのCys431へと,チオエステル結合の形で受け渡される,3)RING2上のユビキチンが,不良ミトコンドリア外膜タンパク質やユビキチンのリシン残基へとイソペプチド結合の形で受け渡される,という三段階からなる(図4A).しかし,自己阻害型parkinの結晶構造では,RING1のE2結合領域にはUBLとREPが,RING2の活性中心Cys431にはUPDがそれぞれ結合しており,周囲からのアクセスが妨げられた状態になっていた(図4B左,C上段)19, 20).また,UBLのSer65はRING1と結合している段階でも溶媒側に露出しているが,RING1とPINK1はUBLの同じ領域に結合するため,PINK1はUBLと結合することができず自己阻害型parkinのリン酸化効率は低い.このように自己阻害型parkinはユビキチンリガーゼ活性が低いだけでなく,PINK1によるリン酸化を受けづらい状態である理由が,構造から示された.
parkinのUPDとRING1上には,リン酸を受け入れる塩基性残基とチロシンによって作られたポケットが存在するが,リン酸化ユビキチンはこのポケットにリン酸化セリン(pSer65)を差し込むように結合する21, 22).リン酸化ユビキチンが結合すると,RING1のC末端ヘリックスが折れ曲がった状態からまっすぐな状態へと変化し,IBRの位置もリン酸化ユビキチンの方へと向かって大きく移動する.この構造変化によりparkinとリン酸化ユビキチンとの結合は増強される.一方,UBLの周りに存在していたIBRおよびIBRとREP間のリンカー領域がUBLから離れるため,UBLとRING1との結合は減弱する(図4C中段).リン酸化ユビキチン結合状態のparkinは,RING2の活性中心がUPDに塞がれている他,RING1とREPの結合も残っているため,活性化状態ではない.しかし,UBLがRING1から解離することで,PINK1によるリン酸化を受けやすい状態になる.PINK1によりUBLのSer65がリン酸化されると,UPD上の塩基性残基からなるポケットに,UBLのpSer65が差し込まれるように結合し,RING2は解離する(図4B右,C下段)23, 24).また,ACTもUPDのRING2相互作用領域と結合することでparkinの活性化に寄与しており,ACTに変異を導入するとparkinの活性は減弱する(ただし,ACTは昆虫では保存されていないため,昆虫の活性型parkinではUBLとUPDの間のループはすべてディスオーダーしていた24)).リン酸化UBLとACTによってRING2がUPDから解離すると,REPもRING1から解離する.おそらくREPがRING1上で固定化されるためには,その隣のドメインであるRING2がUPD上で固定化されている必要があると考えられる.このようなダイナミックな構造変化の結果,RING1のE2結合領域と,RING2の活性中心Cys431は溶媒に露出し,parkinは活性型となる.さらに,この活性型parkinでは,RING2はIBRと長いループ(ヒトでは40残基以上)でつながれているため自由に動くことができるので,E2からRING2へのユビキチンの受け渡しや,RING2から基質(ミトコンドリア外膜タンパク質やユビキチン)へのユビキチンの受け渡しにも適した状態であると考えられる.
このように多くの研究グループによる成果から,parkinの活性化メカニズムは解明されたが,E2~UbからRING2へのユビキチンの受け渡しと,RING2から基質へのユビキチンの受け渡しのメカニズムについてはいまだ解明されていない.また,他のE3では合成されることがほとんどないK6鎖を,parkinがどのように合成しているのかという点は,parkinの機能解明において残された大きな謎である.
本稿で述べたとおり,USP30がK6鎖を特異的に切断するメカニズム,PINK1がユビキチンをリン酸化するメカニズム,ユビキチンのリン酸化がparkinを活性化しUSP30の活性を減弱するメカニズムについては機能・構造解析から明らかとなった.一方,parkinがK6, K11, K48, K63鎖という4種類のユビキチン鎖を伸長するメカニズム,そしてこの4種類のユビキチン鎖のマイトファジーにおける役割の違い,という点についてはほとんどわかっていない.parkinが合成したユビキチン鎖を認識するオートファジーアダプターである,p62, NDP52, OPTNのユビキチン結合領域はユビキチン鎖の種類にかかわらず結合する.しかし,Lys6もしくはLys11をアルギニンに置換してそれぞれのリシン残基でつながったユビキチン鎖が形成されないようにするとマイトファジーが起こらないという報告もあるため,これらの非定型型ユビキチン鎖はそれぞれ違う役割を持っていると考えられる10).さらに,他の経路ではほとんど合成されていないK6鎖をparkinが合成するという点,そしてその下流のUSP30がK6鎖を特異的に切断するという点から,K6鎖はマイトファジーの制御に重要な役割を果たすということが予想されている.K6鎖がマイトファジーを制御する役割については,今後のより詳細な研究が必要である.
引用文献References
1) Kubli, D.A. & Gustafsson, A.B. (2012) Mitochondria and mitophagy: the yin and yang of cell death control. Circ. Res., 111, 1208–1221.
2) Corti, O., Lesage, S., & Brice, A. (2011) What genetics tells us about the causes and mechanisms of Parkinson’s disease. Physiol. Rev., 91, 1161–1218.
3) Pickrell, A.M. & Youle, R.J. (2015) The roles of PINK1, parkin, and mitochondrial fidelity in Parkinson’s disease. Neuron, 85, 257–273.
4) Nakamura, N. & Hirose, S. (2008) Regulation of mitochondrial morphology by USP30, a deubiquitinating enzyme present in the mitochondrial outer membrane. Mol. Biol. Cell, 19, 1903–1911.
5) Narendra, D.P., Jin, S.M., Tanaka, A., Suen, D.F., Gautier, C.A., Shen, J., Cookson, M.R., & Youle, R.J. (2010) PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol., 8, e1000298.
6) Okatsu, K., Oka, T., Iguchi, M., Imamura, K., Kosako, H., Tani, N., Kimura, M., Go, E., Koyano, F., Funayama, M., et al. (2012) PINK1 autophosphorylation upon membrane potential dissipation is essential for Parkin recruitment to damaged mitochondria. Nat. Commun., 3, 1016.
7) Okatsu, K., Uno, M., Koyano, F., Go, E., Kimura, M., Oka, T., Tanaka, K., & Matsuda, N. (2013) A dimeric PINK1-containing complex on depolarized mitochondria stimulates Parkin recruitment. J. Biol. Chem., 288, 36372–36384.
8) Koyano, F., Okatsu, K., Kosako, H., Tamura, Y., Go, E., Kimura, M., Kimura, Y., Tsuchiya, H., Yoshihara, H., Hirokawa, T., et al. (2014) Ubiquitin is phosphorylated by PINK1 to activate parkin. Nature, 510, 162–166.
9) Iguchi, M., Kujuro, Y., Okatsu, K., Koyano, F., Kosako, H., Kimura, M., Suzuki, N., Uchiyama, S., Tanaka, K., & Matsuda, N. (2013) Parkin-catalyzed ubiquitin-ester transfer is triggered by PINK1-dependent phosphorylation. J. Biol. Chem., 288, 22019–22032.
10) Cunningham, C.N., Baughman, J.M., Phu, L., Tea, J.S., Yu, C., Coons, M., Kirkpatrick, D.S., Bingol, B., & Corn, J.E. (2015) USP30 and parkin homeostatically regulate atypical ubiquitin chains on mitochondria. Nat. Cell Biol., 17, 160–169.
11) Bingol, B., Tea, J.S., Phu, L., Reichelt, M., Bakalarski, C.E., Song, Q., Foreman, O., Kirkpatrick, D.S., & Sheng, M. (2014) The mitochondrial deubiquitinase USP30 opposes parkin-mediated mitophagy. Nature, 510, 370–375.
12) Wauer, T., Swatek, K.N., Wagstaff, J.L., Gladkova, C., Pruneda, J.N., Michel, M.A., Gersch, M., Johnson, C.M., Freund, S.M., & Komander, D. (2015) Ubiquitin Ser65 phosphorylation affects ubiquitin structure, chain assembly and hydrolysis. EMBO J., 34, 307–325.
13) Sato, Y., Okatsu, K., Saeki, Y., Yamano, K., Matsuda, N., Kaiho, A., Yamagata, A., Goto-Ito, S., Ishikawa, M., Hashimoto, Y., et al. (2017) Structural basis for specific cleavage of Lys6-linked polyubiquitin chains by USP30. Nat. Struct. Mol. Biol., 24, 911–919.
14) Hospenthal, M.K., Freund, S.M., & Komander, D. (2013) Assembly, analysis and architecture of atypical ubiquitin chains. Nat. Struct. Mol. Biol., 20, 555–565.
15) Gersch, M., Gladkova, C., Schubert, A.F., Michel, M.A., Maslen, S., & Komander, D. (2017) Mechanism and regulation of the Lys6-selective deubiquitinase USP30. Nat. Struct. Mol. Biol., 24, 920–930.
16) Okatsu, K., Sato, Y., Yamano, K., Matsuda, N., Negishi, L., Takahashi, A., Yamagata, A., Goto-Ito, S., Mishima, M., Ito, Y., et al. (2018) Structural insights into ubiquitin phosphorylation by PINK1. Sci. Rep., 8, 10382.
17) Kumar, A., Tamjar, J., Waddell, A.D., Woodroof, H.I., Raimi, O.G., Shaw, A.M., Peggie, M., Muqit, M.M., & van Aalten, D.M. (2017) Structure of PINK1 and mechanisms of Parkinson’s disease-associated mutations. eLife, 6, e29985.
18) Schubert, A.F., Gladkova, C., Pardon, E., Wagstaff, J.L., Freund, S.M.V., Steyaert, J., Maslen, S.L., & Komander, D. (2017) Structure of PINK1 in complex with its substrate ubiquitin. Nature, 552, 51–56.
19) Trempe, J.F., Sauve, V., Grenier, K., Seirafi, M., Tang, M.Y., Menade, M., Al-Abdul-Wahid, S., Krett, J., Wong, K., Kozlov, G., et al. (2013) Structure of parkin reveals mechanisms for ubiquitin ligase activation. Science, 340, 1451–1455.
20) Kumar, A., Aguirre, J.D., Condos, T.E., Martinez-Torres, R.J., Chaugule, V.K., Toth, R., Sundaramoorthy, R., Mercier, P., Knebel, A., Spratt, D.E., et al. (2015) Disruption of the autoinhibited state primes the E3 ligase parkin for activation and catalysis. EMBO J., 34, 2506–2521.
21) Wauer, T., Simicek, M., Schubert, A., & Komander, D. (2015) Mechanism of phospho-ubiquitin-induced PARKIN activation. Nature, 524, 370–374.
22) Kumar, A., Chaugule, V.K., Condos, T.E.C., Barber, K.R., Johnson, C., Toth, R., Sundaramoorthy, R., Knebel, A., Shaw, G.S., & Walden, H. (2017) Parkin-phosphoubiquitin complex reveals cryptic ubiquitin-binding site required for RBR ligase activity. Nat. Struct. Mol. Biol., 24, 475–483.
23) Gladkova, C., Maslen, S.L., Skehel, J.M., & Komander, D. (2018) Mechanism of parkin activation by PINK1. Nature, 559, 410–414.
24) Sauve, V., Sung, G., Soya, N., Kozlov, G., Blaimschein, N., Miotto, L.S., Trempe, J.F., Lukacs, G.L., & Gehring, K. (2018) Mechanism of parkin activation by phosphorylation. Nat. Struct. Mol. Biol., 25, 623–630.
著者紹介Author Profile
 佐藤 裕介(さとう ゆうすけ)
佐藤 裕介(さとう ゆうすけ)鳥取大学工学部附属グリーン・サスティナブル・ケミストリー研究センター講師.博士(理学).
略歴1982年東京都生まれ.2005年東京工業大学生命理工学部卒業.09年同大学院生命理工学研究科博士課程修了.09年より東京大学放射光連携研究機構助教,同大分子細胞生物学研究所助教,同大定量生命科学研究所助教を経て,19年より現職.
研究テーマと抱負タンパク質複合体の立体構造決定により,ユビキチン鎖による細胞機能制御の分子機構の解明をめざしている.
趣味時々絵を描く.