嚢胞性線維症(cystic fibrosis:CF)はcystic fibrosis transmembrane conductance regulator(CFTR)の遺伝子変異に伴う機能喪失を原因とする致死性の劣性遺伝病である.CFは白人種間に高頻度(約3500人に1人)で発症し,世界での患者数は約8.5万人にものぼる(米国:約3万人,欧州:約4万人).日本においては患者数50人未満の希少疾患(指定難病)であり,白色人種とは遺伝的背景を異にする1).CF患者は呼吸器,消化器,外分泌器官(膵臓,精巣,汗腺など)など全身で進行性の病態を呈する.生後まもなくから腸管内腔水分量の低下や慢性膵炎による膵臓からの消化酵素の分泌量低下による胎便性イレウスが生じる.また,呼吸器では気道粘液の粘稠度が増加することで気道クリアランスが低下し,緑膿菌等の細菌感染が持続する.感染による慢性炎症は気道のリモデリング,線維化を促進し,次第に呼吸機能が低下することで致死的な呼吸機能不全に至る.抗生物質や膵消化酵素,去痰薬,抗炎症薬などの対症療法の発展によりCF患者の寿命は延長されたがいまだに余命の中央値は約40歳であり,有効な治療法の確立が求められている.
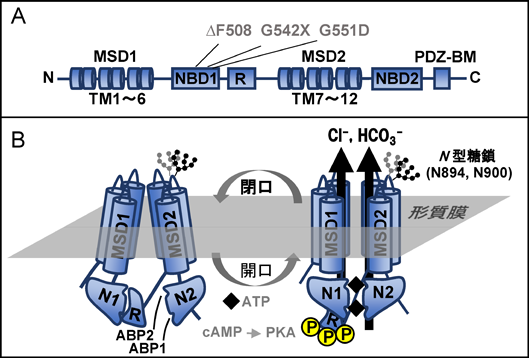
CFTRは上皮細胞のアピカル形質膜に発現するcyclic adenosine monophospate(cAMP)依存性の塩素イオン(Cl−)チャネルであり,重炭酸イオン(HCO3−)輸送にも関わっている2).CFTRは二つの6回膜貫通ドメイン(membrane spanning domain 1, 2:MSD1, MSD2)と制御ドメイン[Regulatory(R)ドメイン],そして二つのヌクレオチド結合ドメイン(nucleotide binding domain 1, 2:NBD1, NBD2)から構成される(図1).CFTRはcAMPによって活性化されたprotein kinase A(PKA)により,Rドメインがリン酸化される.CFTRのRドメインのリン酸化はadenosine triphosphate(ATP)介在性のNBD1, NBD2の二量体形成を生じ,ATP加水分解を駆動力としたCl−,HCO3−の輸送が可能となる3).細胞外へのCl−輸送はトランスポーターまたは傍細胞経路を介したNa+の輸送を生じ,浸透圧効果による水分の管腔側への輸送を誘導するため,CFTRのチャネル機能は細胞外水分量の調節に重要である.また,CFTRにより輸送されるHCO3−は細胞外pHの低下を防ぎ,粘液粘稠化の抑制や細菌感染防御に寄与すると考えられている4, 5).
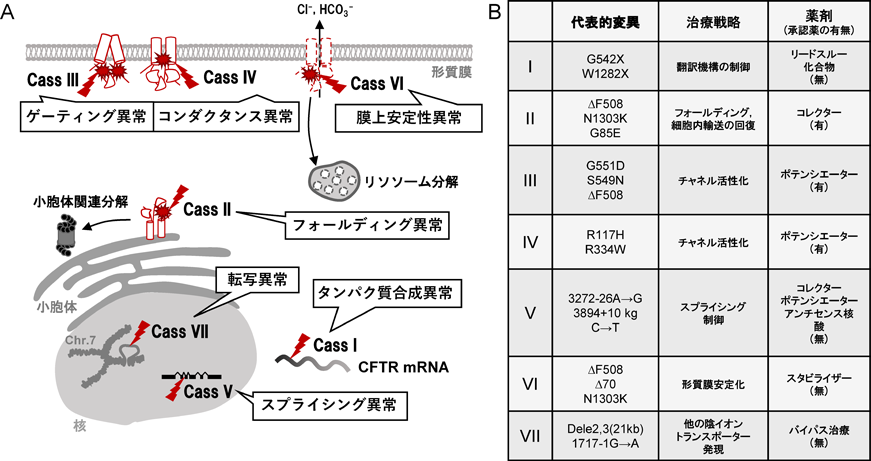
CFTRの変異は現在までに2000種類以上同定されているが,CF患者で最も多い変異はNBD1に存在する508番目のフェニルアラニンが欠失した∆F508変異である.約90%のCF患者が一つの∆F508アレルを有し,約50%のCF患者が∆F508ホモ接合体である(Annual Data Report 2017 Cystic Fibrosis Foundation Patient Registry https://www.cff.org/Research/Researcher-Resources/Patient-Registry/2017-Patient-Registry-Annual-Data-Report.pdf).CFTRの変異はその特性によってClass I~VIIに分類される6)(図2).Class I変異は未成熟終止コドンが生じることで全長のCFTRタンパク質が合成されず,機能的なCFTRタンパク質発現が損なわれる.Class II変異は小胞体におけるタンパク質フォールディングが正常に行われず,ミスフォールドタンパク質として小胞体品質管理機構(endoplasmic reticulum quality control:ERQC)に認識され,プロテアソーム分解される.そのため,プロセッシング(成熟化)とトラフィッキング(細胞内輸送)に異常を生じ,形質膜発現が失われる.Class IIIおよびIV変異はCFTRタンパク質の成熟化および形質膜発現には影響を与えないが,イオンチャネル機能を喪失もしくは低下してしまう.Class V変異は転写やスプライシング異常によってCFTRの発現量の変化を生じる.Class VI変異はCFTRタンパク質の形質膜への移行,およびチャネル活性には影響を与えないが,形質膜上での不安定化を引き起こす結果,形質膜発現を低下させる.Class VII変異は転写異常によってCFTR mRNAが産生されない変異型である.先述した∆F508変異はフォールディング異常を示すClass II変異であると考えられていたが,近年,Class IIIおよびVI変異としての性質も有することが明らかにされている7, 8).
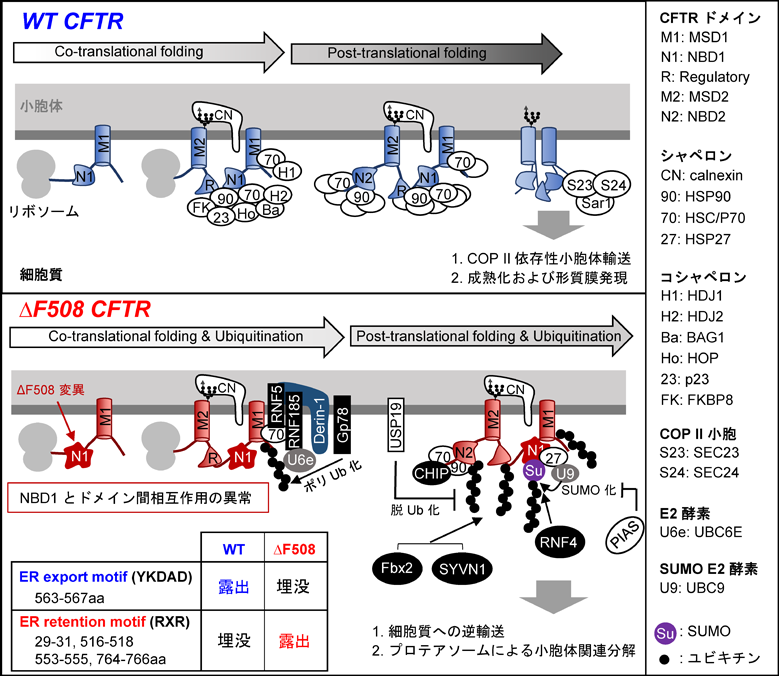
CFTRは小胞体膜上で翻訳され,正しい立体構造へとフォールディングする.CFTRフォールディング機構は,各ドメインが翻訳と同時に準安定状態までフォールディングする第一段階(co-translational domain-wise folding)と翻訳後に各ドメインが相互作用し,CFTR全体として天然状態にフォールディングする第二段階(post-translational coupled-domain folding and assembly)の少なくとも二段階で起こると考えられている9–12).CFTRは小胞体でN型糖鎖修飾を受け,小胞体膜貫通型レクチン様シャペロンであるcalnexinと相互作用する13, 14).また,CFTRは細胞質分子シャペロンであるheat shock protein 70(HSP70),HSP90およびその補助因子であるコシャペロン(HDJ2, p23, FKBP8)とも相互作用し,そのフォールディングが補助される15–17)(図3).
CFで最も多いΔF508変異はNBD1に存在する508番目のフェニルアラニン(F508)が欠失する変異であり,NBD1の構造安定性を低下させる18).また,F508はNBD1の表面に位置し,NBD1とMSD1およびMSD2の細胞内ループとの境界に位置するため,ΔF508変異はNBD1とMSD1/2のドメイン間相互作用も不安定化すると考えられている11, 19).実際に,NBD1構造安定性を改善する点変異(solubilizing mutation)はΔF508 CFTRフォールディングを部分的にしか改善できないが,NBD1とMSD1/2のドメイン間相互作用を安定化するR1070W変異を併用することで,ΔF508 CFTRフォールディングがほぼ完全に改善される19, 20).したがって,ΔF508変異によるCFTRフォールディング異常を改善するためには,NBD1構造不安定性とNBD1とMSD1/2のドメイン間相互作用の両方を同時に改善する必要があると考えられる9).
翻訳速度もCFTRフォールディング効率に影響を与える21).リボソームタンパク質ribosomal protein L12(RPL12)ノックダウンはCFTR変異体の翻訳速度を低下させる結果,ΔF508 CFTRフォールディングが部分的に改善することが報告されている22).実際に,低濃度のcycloheximideやemetineにより新生ポリペプチド伸長を減速させると,ΔF508 CFTRのフォールディングの改善が観察されている22).
1)小胞体品質管理機構
小胞体において,ミスフォールドしたΔF508 CFTRは小胞体品質管理機構により,小胞体に滞留(ER retention)される(図3).CFTRの小胞体からの小胞輸送にはcoat protein complex(COP)II小胞コートタンパク質SEC23/SEC24複合体により認識されるdi-acidic ER export motifが関与し,ΔF508 CFTRではフォールディング異常によりこの小胞体輸送シグナルが露出しないため,小胞体に滞留される可能性が考えられている23).また,ΔF508 CFTRの小胞体滞留にはRXR-based ER retention/retrieval motifsが関与し,フォールディング異常によりこの小胞体滞留シグナルが露出されると考えられている24, 25).小胞体に滞留されたΔF508 CFTRは細胞質のheat shock cognate 70(HSC70)やHSP70,小胞体のcalnexinといった分子シャペロンとの相互作用が持続する14, 26–29).小胞体に滞留されたΔF508 CFTRは最終的にユビキチン化を受け,Derlin-1複合体を介して細胞質へ逆行輸送された後,プロテアソームで分解される30–32).
小胞体におけるΔF508 CFTRのユビキチン化には多くのユビキチンリガーゼが関与する.CFTRのユビキチン化は,翻訳と同時に起こるco-translational ubiquitinationと翻訳後に起こるpost-translational ubiquitinationが考えられている30, 33).ring finger protein 5(RNF5/RMA1)は小胞体膜上に存在するRING型ユビキチンリガーゼであり,MSD1などのCFTRの膜貫通領域を主に認識すると考えられており,co-translational ubiquitinationへの関与が示唆されている34).RNF185はRNF5ホモログであり,小胞体膜上に存在するRING型ユビキチンリガーゼである.RNF5と同様にDerlin-1, ubiquitin conjugating enzyme 6E(UBC6E)と相互作用し,ΔF508 CFTRのco-translational ubiquitinationへの関与が示唆されている35).RNF5とRNF185の機能は一部重複する一方,異なる機能も示唆されているが,機能的な違いは不明である.Gp78(AMFR)は小胞体膜上に存在するRING型ユビキチンリガーゼであり,ΔF508 CFTRの小胞体関連分解に関与するGp78はE4様活性があり,RNF5によるCFTRのユビキチン化を伸長すると考えられている36).carboxy terminus of Hsp70-interacting protein(CHIP)は細胞質に存在するシャペロン結合性ユビキチンリガーゼであり,ΔF508 CFTRの細胞質領域,特に,NBD2が合成された後の翻訳後ユビキチン化(post-translational ubiquitination)に関与すると考えられている29, 34).これら以外のユビキチンリガーゼであるSkp1-Cullin1-Fbx2-Roc1 ubiquitin ligase complex(SCFFbx2)やsynoviolin 1(SYVN1)の関与も報告されているが,ミスフォールドしたΔF508 CFTRを選択的に認識し,ユビキチン化する証拠が不足しており,CFTR小胞体品質管理に関与するか否かは不明である37, 38).小胞体におけるCFTRのユビキチン化は可逆的であり,小胞体膜tail-anchored脱ユビキチン化酵素(deubiquitinating enzyme:DUB)であるubiquitin specific peptidase 19(USP19)はΔF508 CFTRのユビキチン化レベルを減少させることで,小胞体関連分解を抑制すると考えられている39).
CFTRはユビキチン化だけではなく,ユビキチン様タンパク質であるSUMOによる修飾(SUMO化)も受ける.small heat shock protein(sHsp)であるHSP27はΔF508 CFTRを選択的に認識し,SUMO E2酵素UBC9と協調してΔF508 CFTRをSUMO化する.SUMO化されたCFTRはSUMO-targeted Ub E3 ligaseであるRNF4によりユビキチン化され,プロテアソーム分解を受ける40).また,HSP27-UBC9複合体は構造異常を持つΔF508-NBD1のK447を選択的にSUMO化し,特にSUMO-2修飾が顕著である41).SUMO E3であるprotein inhibitor of activated STAT4(PIAS)はCFTRのSUMO-1修飾を促進し,SUMO-2/3修飾を阻害する結果,CFTRのユビキチン化を阻害し,その分解を抑制する42).
2)形質膜タンパク質品質管理機構
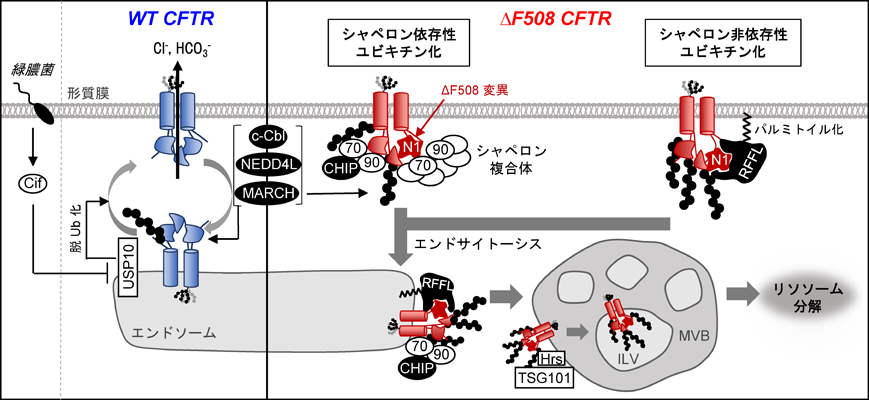
ΔF508 CFTRの大部分は小胞体関連分解で除去されるが,一部のΔF508 CFTRは形質膜に移行する.実際に,CF患者の気道上皮細胞および腸管上皮細胞において,ΔF508 CFTRのわずかな形質膜発現が観察されている43).ΔF508 CFTRのフォールディングは温度感受性であり,低温培養(26~30°C)によりΔF508 CFTRの形質膜発現は促進される44).また,CF治療薬の主成分であるCFTRコレクターもΔF508 CFTRの形質膜発現を促進する45).形質膜に出現したΔF508 CFTRはイオンチャネルとして機能できるが,構造異常タンパク質としてユビキチン化され,速やかに分解される46–48)(図4).また,C末端が欠損したΔ70 CFTRなどのClass VI変異体は,成熟化および形質膜への移行は正常に起こるが,形質膜でユビキチン化を受け,速やかに分解される46).ユビキチン化はCFTRのエンドサイトーシスを促進し,リサイクリングを阻害する.ユビキチン化されたCFTRはエンドソームでHrs, TSG101を含むendosomal sorting complex required for transport complex(ESCRT)複合体に認識された結果,リソソーム分解される46, 49).形質膜からのCFTR変異体の分解にはプロテアソームも関与するが,これらの異なる分解経路の仕分け機構はいまだ不明である50, 51).
形質膜におけるΔF508 CFTRのユビキチン化は,シャペロン依存性ユビキチンリガーゼCHIPが関与する.形質膜のΔF508 CFTRは部分的に変性しており,その構造異常はHSC70, HSP90とコシャペロンであるDnaJ heat shock protein family member A1(DNAJA1),HSP70-HSP90 organizing protein(HOP)やactivator of HSP90 ATPase protein 1(AHA1)などに認識される49).これらのシャペロン複合体は形質膜のΔF508 CFTRの構造安定性およびチャネル機能の維持に重要である52).しかしながら,構造異常を持つ形質膜のΔF508 CFTRは小胞体品質管理機構と同様に,分子シャペロンとの結合が持続し,最終的にCHIPがHSC70, HSP90と結合することで,ΔF508 CFTRのユビキチン化が起こると考えられる49, 53).形質膜ΔF508 CFTRのユビキチン化にはシャペロン非依存性ユビキチンリガーゼring finger and FYVE like domain containing E3 ubiquitin protein ligase(RFFL)も関与することが近年明らかとなった54).RFFLはN末端領域にPI(3)P, PI(5)Pと結合するFYVE-like domain, C末端領域にユビキチンリガーゼ活性に重要なRING domainを有し,それ以外の領域に天然変性領域(disordered region)を多く含む54, 55).RFFLは膜貫通領域を持たないが,N末端領域がパルミトイル化され,形質膜およびエンドソーム膜の細胞質側に局在する56, 57).RFFLはN末端領域の天然変性領域を介して,ΔF508 CFTRの細胞質領域NBD1を直接認識する.興味深いことに,RFFLによる認識はNBD1の構造依存的であり,部分的に変性したNBD1を選択的に認識して,ユビキチン化する.RFFLノックダウンは小胞体に局在するΔF508 CFTR未成熟型のユビキチン化および小胞体関連分解にはほとんど影響しないが,形質膜やエンドソームに局在する成熟型ΔF508 CFTRのユビキチン化,特に,リソソーム分解に深く関与するK63連結型ポリユビキチン化を阻害し,形質膜からのリソソーム分解を抑制する54).したがって,RFFLは形質膜およびエンドソームに局在するΔF508 CFTRの構造異常を選択的に認識し,分解へ運ぶ形質膜品質管理ユビキチンリガーゼとして機能すると考えられる.CHIP, RFFL以外にも,ユビキチンリガーゼMARCH2, c-CBLおよびNEDD4Lが形質膜およびpost–Golgi区画におけるCFTR分解に関与する報告があるが,構造異常を有するCFTRを選択的に制御する証拠が不足しており,これらのユビキチンリガーゼがCFTR品質管理に関与するかは不明である58–61).
小胞体品質管理機構と同様に,CFTR形質膜品質管理機構にDUBが関与する可能性が高い.エンドソーム局在DUBであるUSP10は初期エンドソームにおいて野生型CFTRのユビキチン化レベルを抑制し,CFTRの形質膜へのリサイクリングを促進する62).CF患者や慢性閉塞性肺疾患(COPD)患者で感染がみられる緑膿菌の病原性因子CifはUSP10活性を抑制し,CFTRのユビキチン化およびリソソーム分解を促進する63).したがって,USP10はエンドソームにおいてCFTRの構造状態を認識し,そのユビキチン化レベルを制御する可能性が高い.しかしながら,CFTRの構造状態を認識し,選択的に天然状態のCFTRを脱ユビキチン化するDUBはいまだ同定されていない.
近年,CFの根本的治療薬としてCFTRを標的とした分子標的薬(CFTR modulator)が開発されている.現在までに,小胞体でのCFTRフォールディングを改善し,形質膜発現を促進するCFTRコレクター,イオンチャネル機能を改善するCFTRポテンシエーター,形質膜安定性を向上させるCFTRスタビライザー,CFTRタンパク質合成を増強するCFTRアンプリファイヤーの4種類が開発されている(図2B).
CFTRポテンシエーターIvacaftor(VX-770, Kalydeco®)は初めてのCF根本的治療薬として2012年に欧米で上市され,G551DなどCFTR Class III変異を有する約10%のCF患者に使用されている.IvacaftorはCFTRの膜貫通領域に直接的に結合し,チャネル開口状態を安定化することで,開口確率を改善すると考えられている64).
Lumacaftor(VX-809)は臨床適用された初めてのCFTRコレクターであり,フォールディング異常により小胞体関連分解を受けるCFTR Class II変異体の治療に用いられる.臨床試験において,Lumacaftor単剤では有効な治療効果が得られなかったため,LumacaftorはCFTRポテンシエーターIvacaftor(VX-770)との配合薬Orkambi®として使用される.Orkambi®はCFの大部分を占める∆F508 CFTR変異を有するCF初の治療薬として2015年に米国食品医薬品局(FDA)に認可を受けた.Lumacaftorは薬物代謝酵素CYP4を強く誘導し,Ivacaftorの血中濃度を大きく減少させるため,薬物相互作用を改善したCFTRコレクターTezacaftor(VX-661)がLumacaftorのアナログとして開発されている65).TezacaftorとIvacaftorの配合薬Symdeco®も2018年からCF根本的治療薬として臨床適用されている.LumacaftorはMSDのフォールディング改善効果に加え,MSD1/2とNBD1のドメイン間相互作用を改善することでCFTR構造を安定化させる66–69).しかしながら,∆F508 CFTRフォールディングの回復に必須なNBD1の安定性改善効果は認められていない69).実際に,Orkambi®,Symdeco®ともに臨床での治療効果は弱く,有効性は疑問視されている70).Kalydeco®と同様に,Orkambi®,Symdeko®は非常に高価で,1年間の治療費はともに2000万円以上であり,その費用対効果を懸念して英国立医療技術評価機構(NICE)は英国民保健サービス(NHS)でのOrkambi®の使用を推奨しないと発表している71).
近年,Lumacaftor, Tezacaftorと異なる作用機序を持つ第2世代CFTRコレクターとしてElexacaftor(VX-445)が開発された.Elexacaftorは第1世代CFTRコレクター(Lumacaftor, Tezacaftor)との併用により∆F508 CFTR膜発現を増加させることから,第1世代CFTRコレクターとは作用点が異なると考えられる72).第一世代コレクターの有効性はNBD1安定化効果により相乗的に増強されることから,ElexacaftorはNBD1安定化作用を有する可能性が考えられる69).Elexacaftor, TezacaftorおよびIvacaftorの3種配合薬は2019年10月にTrikafta®としてFDA認可を受けた.Symdeco®は4週間投与により呼吸機能を示す対標準一秒量(%FEV1)を約7%改善する一方,Trikafta®は約14%の改善効果がみられることから,臨床での高い有効性が期待されている72, 73).
CFTRアンプリファイヤーはCFTRの転写・翻訳機構に作用し,CFTRタンパク質量を増加させることを企図した薬剤であり,他のCFTR modulatorとの併用により相乗作用が期待される74).PTI-428はいまだ機序に不明な点があるがCFTRのmRNA安定性の向上もしくはCFTR mRNAの翻訳機構に作用し,小胞体で合成されるCFTRタンパク質量を増加するとされる75).PTI-428はTezacaftorおよびIvacaftorと併用することによってCFTR機能を約2倍まで増加させることが∆F508ホモ接合型変異患者の第II相臨床試験で示されている(NCT02718495).HDAC阻害剤である4-phenylbutyrate(4-PBA)は非特異的にCFTR mRNAレベルを増加し,CFTR変異体の発現・機能を改善すると考えられる74–76).histone deacetylase 7(HDAC7)阻害剤SAHA1やromidepsin(FK-228)はCFTRの合成,フォールディング,細胞内小胞輸送,品質管理に関わる多くの分子の遺伝子発現をエピゲノム的に変化させる結果,∆F508 CFTRなどのCFTR変異体の発現・機能を改善すると考えられている77–79).
CFTRスタビライザーは変異CFTRに生じる形質膜不安定性を改善することで形質膜安定性を高め,CFTR膜発現および機能の持続を促す薬剤である.形質膜不安定性を示すClass VI変異やCFTRコレクターにより形質膜に発現した∆F508 CFTRは,形質膜においてその構造の不安定性によりシャペロン依存的,もしくは非依存的にユビキチン化を受ける.ユビキチン化されたCFTRは形質膜もしくはエンドソームにおいてポリユビキチン化され,リソソーム分解経路へと送られる.正常型CFTRの形質膜半減期が15時間以上であるのに対し,∆F508 CFTRでは約2時間程度と短い46, 48, 49, 80).Cavosonstatは初めて臨床試験が行われたCFTRスタビライザーであり,変異CFTR形質膜不安定化を誘導するシャペロン調節因子HOPのS-ニトロソ化を促進することで変異CFTRとHOPの相互作用を阻害し,CFTR形質膜安定性を改善する可能性が示唆されている81).しかしながらCavosonstatは臨床(第II相)試験で有効性が示されず,認可には至っていない.CFTRポテンシエーターによってCFTR形質膜安定性が低下すること,また第二世代CFTRコレクターを用いた3剤配合薬でもΔF508 CFTR形質膜安定性は野生型レベルまで回復しないことからも,CFTRスタビライザーの開発は急務である82–84).
5. CFTR品質管理機構を標的としたCF治療戦略
多くのCFにおいて,CFTR機能欠損はCFTR変異体の分解により引き起こされることから,CFTR分解阻害がCF根本的治療に有効であると考えられる.しかしながら,プロテアソーム阻害剤処理はΔF508 CFTRの小胞体関連分解を抑制するが,細胞質にユビキチン化ΔF508 CFTRを蓄積させ,アグリソームを形成する85).一方で,小胞体分子シャペロンcalnexin過剰発現によりΔF508 CFTRのユビキチン化および細胞質への逆輸送(retro-translocation)が阻害されると,形質膜に発現可能なfoldable ΔF508 CFTRが小胞体膜上に蓄積する14).ユビキチン化はΔF508 CFTRの逆輸送を促進すると考えられることから86),CFTR選択的なユビキチン化の阻害,つまり,基質選択性を担うCFTR関連ユビキチンリガーゼの阻害は,形質膜に発現可能なfoldable ΔF508 CFTRを蓄積させ,治療効果が弱いCFTRコレクター薬効の増強に有効であると考えられる.実際に,細胞株モデルにおいて,ΔF508 CFTRの小胞体関連分解に関与するユビキチンリガーゼRNF5, CHIP, SYVN1のノックダウンはCFTRコレクターの薬効を増強する38, 87).マウスモデルにおいても,RNF5ノックアウトがΔF508 CFTR発現・機能を改善し,CF病態を改善することが示されている88).また,RNF5低分子阻害剤(Inh-02)は前臨床試験の最終評価系であるCF患者気道上皮初代培養細胞モデルにおいて,ΔF508 CFTR発現および機能を改善する89).したがって,CFTRユビキチンリガーゼ阻害剤はCF根本的治療薬,もしくはその有効性を増強するアドオン薬として有効であると考えられる.CFTR形質膜品質管理に関与するRFFLはΔF508 CFTRと直接的に結合すること,さらに,RFFLノックアウトマウスが健常であることから,CF治療標的として有効である可能性が高い54, 90).RFFLノックダウンがΔF508 CFTR形質膜安定性を改善することからも,RFFL阻害剤はfirst-in classのCF根本的治療薬であるCFTRスタビライザーになる可能性が期待される91).