受容体型チロシンキナーゼによる細胞分裂制御Regulation of cell division by receptor-type tyrosine kinase
京都薬科大学Kyoto Pharmaceutical University ◇ 京都市山科区御陵中内町5 ◇ 5 Misasaginakauchi-cho, Yamashina-ku, Kyoto
発行日:2020年4月25日Published: April 25, 2020
© 2020 公益社団法人日本生化学会© 2020 The Japanese Biochemical Society
細胞分裂は,複製したDNAを二つの娘細胞に分配する過程である.セリン/トレオニンキナーゼであるCDK1が種々のタンパク質をリン酸化すると,染色体の凝縮,核膜崩壊,中心体の分離などにより細胞分裂が開始する.中心体から伸びた微小管が染色体を捕捉し,細胞の赤道面上に染色体を並べる.すべての染色体のキネトコアに両極からのびた紡錘糸が正しく結合すると,紡錘体チェックポイントが解除され,染色体が分配する細胞分裂後期が始まる.分離した染色体の間には,微小管の束が逆並行に相互作用しミッドゾーンを形成する.ミッドゾーンにはAurora Bキナーゼなどが局在し,セントラルスピンドリンなどの下流分子に作用し,その後の細胞質分裂を制御する.赤道面の細胞膜直下にアクトミオシンにより構成される収縮環が形成して収縮し,ミッドボディー付近が切り離されて細胞は二つに分離する.
この非常にダイナミックな過程をオーガナイズするのが,CDK1,Auroraキナーゼ,Polo-like kinase(PLK)などのセリン/トレオニンキナーゼである.これまで我々は,非受容体型チロシンキナーゼであるSrc型チロシンキナーゼによる細胞分裂制御を報告してきたが,チロシンキナーゼによる細胞分裂制御の報告は少ない.特に,受容体型チロシンキナーゼによる細胞分裂制御に関する報告はきわめて少ない.本稿では,最近我々が報告した,細胞分裂におけるEphA2チロシンキナーゼの役割を中心に説明する1).
Eph受容体は細胞膜上に存在する受容体型チロシンキナーゼであり,受容体型チロシンキナーゼの最も大きなファミリーを形成している2, 3).アミノ酸配列とそのリガンドとの親和性から,EphA,EphBの二つのサブクラスに分類される.それぞれのリガンドであるephrin Aおよびephrin Bは,それぞれGPIアンカー型タンパク質および膜貫通型タンパク質であることから,受容体への結合には細胞間接着が必要である.そのシグナルにより,細胞骨格のリモデリング,細胞の増殖や分化など,さまざまな細胞機能に関与する.また,リガンド/受容体結合により誘導される細胞-細胞間の反発作用は,組織形成過程における細胞移動や領域形成に関与する.EphA2はEph受容体ファミリーメンバーの一つであり,そのリガンドephrin A1と結合するとキナーゼ活性が亢進し,傍膜貫通領域のTyr588とTyr594などがリン酸化する(図1).一方,EphA2が過剰発現しているがん細胞においては,リガンド刺激には依存せず,EGF,HGF,TNFαなどの増殖因子の刺激によりSer897のリン酸化が誘導され,細胞の運動性を促進してがんを悪性化させることが知られている4–7).我々は,ケミカルジェネティクスの手法により,細胞分裂を制御する新たな分子の発見とその制御の解明を目指している.その中で,受容体型チロシンキナーゼファミリーに属するEph受容体の阻害剤が細胞分裂進行を遅延させることを見いだした1).

チロシンキナーゼであるEphA2受容体は膜貫通ドメインを持つ膜内在性タンパク質であり,リガンドephrin A1はGPIアンカーにより細胞膜に係留している.EphA2受容体は以下のドメイン構造,モチーフを持つ:リガンド結合ドメイン,Sushiドメイン,EGF様ドメイン,フィブロネクチンドメイン,膜貫通ドメイン,傍膜貫通領域,キナーゼドメイン,SAMドメイン,PDZドメイン結合モチーフ.細胞-細胞間接着によりEphA2がephrin A1と相互作用すると,チロシン残基(Tyr588, Tyr594, Tyr772)の自己リン酸化が起こる(リガンド依存).一方,増殖因子やTNFα刺激が,Akt,PKA,RSKキナーゼの活性化を介しSer897をリン酸化する.このシグナル経路にはephrin A1との結合は必要ない(リガンド非依存).
細胞分裂の進行を評価する方法として,CDK1の可逆的な阻害剤RO-3306が有用である8).RO-3306により細胞周期をG2/M期境界に止め,これを除いて細胞分裂開始を誘導し,継時的に細胞を固定して染色体・微小管の形態に基づき細胞分裂の進行を評価する.Eph受容体阻害剤NVP-BHG712の存在下で細胞分裂を開始させると細胞分裂進行が遅延したことから,Eph受容体の細胞分裂への関与が示唆された.このとき,EphA2のキナーゼ活性の指標となるチロシン残基(Tyr588)のリン酸化が低下したため,EphA2のキナーゼ活性が細胞分裂制御に関与すると当初は考えた.
一般的に阻害剤の標的分子は単一ではない.そこでsiRNAによりEphA2を発現抑制し細胞分裂への影響を調べると,細胞の中心に位置しない紡錘体や多極性紡錘体が形成するとともに細胞分裂後期への移行が遅延し,この細胞分裂遅延は野生型EphA2の再発現により抑制された.これらの表現型はhTERTにより不死化した網膜色素上皮細胞株hTERT RPE-1細胞やトリプルネガティブ乳がん細胞株MDA-MB-231細胞においても観察された.よって,EphA2を発現する細胞において,EphA2による細胞分裂制御が存在することが示唆された.
細胞分裂の進行にEphA2の関与が示されたので,チミジンを用いて細胞分裂期とG2期後半に同調しTyr588のリン酸化を調べた.G2期の細胞でこのリン酸化が検出されたのに対し,細胞分裂期では検出されなかった.受容体型チロシンキナーゼが活性化するためには,リガンドとの結合が必要である.多くの培養細胞は,細胞分裂において細胞間接着が減弱し球状に形態変化する.EphA2のリガンドであるephrin A1は膜結合型であるため,細胞–細胞間接着を失うとリガンドとの結合も失われると考えられる.したがって,細胞分裂期でのTyr588のリン酸化低下は妥当である.このリン酸化の消失は,キナーゼ活性の消失を示している.EphA2をノックダウンし,野生型あるいはキナーゼ活性不活化変異体を発現させると,野生型のみならずキナーゼ活性不活性化変異体の再発現によっても細胞分裂遅延が解除された.したがって,EphA2による細胞分裂制御にキナーゼ活性は関与しないことが明らかになった(投稿中).
EphA2は増殖因子やTNFαの刺激によりSer897がリン酸化され,キナーゼ活性に依存せず細胞の増殖,遊走に関与することが知られている.チミジンあるいはRO-3306を用いた同調により細胞分裂期の細胞を集め調べると,細胞分裂期ではTyr588ではなくSer897のリン酸化が観察され,細胞分裂が後期/終期に進行するとこのリン酸化は低下した.細胞分裂のマスターレギュレーターであるCDK1は細胞分裂中期から後期へ移行する前に不活性化する.したがって,Ser897のリン酸化の変化はCDK1の活性の変化と一致している(図2).恒常的活性化型CDK1(T14A/Y15F)変異体と分解を抑制したcyclin B1の間期細胞への導入によりSer897がリン酸化されたことは,CDK1の下流でEphA2がリン酸化されること,すなわち,細胞分裂開始時のCDK1活性に依存してEphA2のSer897がリン酸化されることを支持している.間期から細胞分裂期への移行によりTyr588リン酸化からSer897リン酸化へ完璧なスイッチが起こる.細胞間接着が消失し球状化することによりTyr588リン酸化を失うことは妥当だが,積極的にそのTyr588リン酸化を抑制するような機構の存在も考えられ興味深い.
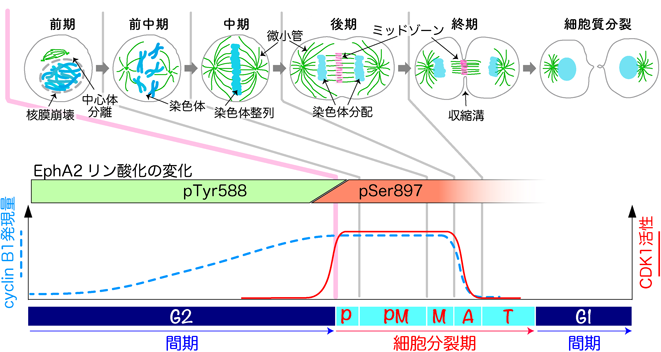
細胞分裂は核膜崩壊,中心体分離,染色体形成などの形態変化により開始し(前期,P),微小管が細胞の赤道面上に染色体を整列させる(前中期,PM).整列したすべての染色体の動原体に両極から伸びた微小管が適切に結合すると(中期,M),紡錘体形成チェックポイントが解除され染色体が分配する(後期,A).分配中の染色体の間で,方向性が逆の微小管が相互作用しミッドゾーンを形成する.染色体が中心体近傍まで移動し,染色体の脱凝縮と核膜の再構成が起こり細胞分裂は終了する(終期,T).細胞質は後期/終期において形成される収縮環により二つの娘細胞に分離する(細胞質分裂).細胞周期間期ではEphA2のTyr588のリン酸化が検出されるが,細胞分裂期ではSer897のリン酸化が検出され,細胞分裂後期になると消失する.cyclin B1はG2期に蓄積し,細胞分裂中期から後期への移行時に分解する.CDK1は細胞周期に依存せず発現しているが,cyclin B1との結合や抑制性のリン酸化修飾の脱リン酸化などにより活性化し細胞分裂が開始する.細胞分裂中期に紡錘体チェックポイントが解除されるとcyclin B1が分解し,CDK1は不活性化して細胞分裂が終了する.すなわち,Ser897がリン酸化されるタイミングはCDK1活性と一致する.
間期ではAKT,PKA,RSKがEphA2のSer897をリン酸化することが知られており(図1参照)4–7),細胞分裂期におけるEphA2-Ser897のキナーゼを阻害剤を用いて探索した.その結果,MEK/ERK阻害剤,RSK阻害剤によりこのリン酸化が低下し,一方,PKAやAKTの阻害剤では低下しなかった.同様に,CDK1導入により亢進したSer897リン酸化もMEKやRSKの阻害剤により低下した.したがって,細胞分裂期では増殖因子シグナルではなく,細胞分裂を制御するCDK1の下流でERK,RSKが活性化し,Ser897をリン酸化すること,すなわち,CDK1/MEK/ERK/RSK/EphA2–Ser897経路の存在が明らかになった(図3).
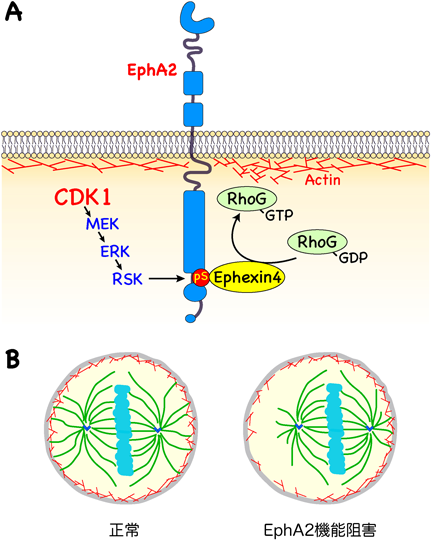
(A)細胞分裂期に,EphA2のSer897がリン酸化されてEphexin4をリクルートし,RhoGをGDP結合型からGTP結合型へ活性化する.RhoGはアクチン線維のリモデリングあるいは細胞膜への結合を制御し,細胞膜の剛性維持に寄与する.(B)細胞膜直下のアクチン線維と微小管との結合は紡錘体の形態や位置にも関与する.EphA2の機能阻害は細胞膜の剛性を低下させることから,細胞膜直下のアクチン線維と微小管の結合の異常が紡錘体の形成や細胞内の位置の異常を引き起こすことが考えられる.
EphA2のSer897のリン酸化は細胞分裂進行においてどのような意味を持つのだろうか.野生型EphA2,あるいはSer897をAlaに変換した変異体(Ser897Ala)を過剰発現する細胞株を樹立し,変異が細胞分裂進行に与える影響を解析した.内在性のEphA2のノックダウンが細胞分裂遅延を引き起こし,野生型EphA2の再発現により回復するが,Ser897Ala変異体では回復しなかったことから,CDK1依存のEphA2-Ser897のリン酸化が細胞分裂進行に必要であることがわかった.キナーゼ活性の阻害剤であるNVP-BHG712によりTyr588に加えSer897のリン酸化の低下も観察された.よって,この阻害剤で観察された細胞分裂遅延は,キナーゼ活性の阻害には起因せず,Ser897リン酸化阻害によることが示唆された.
増殖因子の下流でEphA2のSer897がリン酸化すると,EphA2はRhoGのGEFであるEphexin4をリクルートしRhoGを活性化する9).細胞分裂期におけるこれらのタンパク質間の相互作用を免疫沈降により検討した.Eg5モータータンパク質の阻害剤であるSTLCを用いてMDA-MB-231細胞を細胞分裂期に同調し,内在性Ephexin4を免疫沈降するとEphA2が共沈した.間期と比較し細胞分裂期において共沈する量が増加し,この相互作用はSer897のリン酸化に依存した.Ephexin4をノックダウンすると細胞分裂の進行が遅延したことから,EphA2はSer897リン酸化に依存してEphexin4をリクルートし細胞分裂進行に関与することが示唆された.
Ephexin4の標的であるRhoGは細胞分裂期において細胞膜に局在したが,MEK/ERK阻害やEphexin4のノックダウンによりその膜局在が減少したため,MEK/ERK/EphA2–pSer897/Ephexin4経路によるRhoGの活性化が膜局在に必要である.RhoGノックダウンは細胞分裂遅延を引き起こし,また,EphA2のノックダウンによる細胞分裂遅延は,野生型RhoGの再発現では回復せず,恒常的活性化変異体(RhoG-Gly12Val)により部分的に回復した.すなわち,EphA2-pSer897はEphexin4をリクルートしてRhoGを活性化し細胞分裂制御に関わっている(図3A).
細胞分裂期において,細胞膜直下のアクチン細胞骨格は再構成され,細胞膜の剛性を維持する.アクチン重合阻害剤であるサイトカラシンを添加しタイムラプス解析すると,2 µg/mLの濃度において細胞膜の突出であるブレブ形成がすべての細胞で観察された.0.25 µg/mLに濃度を下げると,ブレブ形成細胞の割合が10~20%程度に低下し,EphA2をノックダウンすると50%程度まで有意に増加した.よって,EphA2は細胞膜の剛性に関与することが明らかになった.
内在性EphA2のノックダウンはブレブ形成を増加し,野生型EphA2の再発現はこれを抑制したがSer897Ala変異体は抑制しなかった.この結果はブレブ形成の抑制にSer897のリン酸化が関与することを示している.さらにMEK/ERK阻害やRhoGノックダウンによってもブレブ形成が増加した.よって,EphA2-pSer897/Ephexin4/RhoGシグナルがアクチン再構成を介して細胞膜直下のアクチン線維の形成,あるいはアクチン線維の細胞膜への結合に関与し,剛性の維持に関わることが示唆された(図3).
細胞膜直下のアクチン線維と微小管との結合は紡錘体の形態や位置にも関与することが知られている10).よってこれらの結果から,CDK1の活性化により細胞分裂が開始すると,CDK1/MEK/ERK/RSK経路によりEphA2のSer897がリン酸化され,Ephexin4をリクルートしてRhoGを活性化する.活性化したRhoGは細胞膜に局在し,細胞膜の剛性維持を介して紡錘体形成に関わり細胞分裂進行に寄与することが示唆される(図3B).EphA2は個体発生時の組織形成に寄与することが知られている.これまで知られていた,リガンド刺激によるキナーゼ活性に依存したシグナルのみならず,細胞分裂制御に関与することで組織形成に関与する可能性が考えられる.がん細胞においてはリガンドであるephrin A1の発現低下がリガンド依存のシグナルを抑制し,Ser897リン酸化シグナルを亢進することも知られているが,正常細胞においては細胞周期に依存してSer897がリン酸化し,リガンド刺激は抑制されることになる.培養細胞の場合は細胞分裂期に球状化することで細胞–細胞間接着を消失し,リガンドであるephrin A1刺激を失うことは想像しやすい.一方,組織形成途中に細胞間接着を保持している細胞で,同様にリガンド刺激を受けないのであれば,どのようにそのリガンド刺激を抑制しているのか今後検討が必要である.
EphA2チロシンキナーゼ以外にも受容体型チロシンキナーゼによる細胞分裂制御が報告されている.受容体型チロシンキナーゼの一つ,インスリン受容体は,インスリンと結合し活性化されるとグルコース取り込み促進やグリコーゲン合成促進により血糖値の低下をもたらす.最近,膵β細胞増殖時の細胞分裂に対するインスリンシグナルの関与が報告された11)インスリン受容体をノックアウトしたβ細胞では,G0/G1期細胞の割合増加,S期,G2/M期の細胞減少,さらにM期遅延が観察された.この細胞ではキネトコアタンパク質であるCENP-Aや細胞分裂制御に関わるキナーゼであるPLK1の発現レベルが低下した.野生型インスリン受容体を導入すると,インスリン刺激によりCENP-A, PLK1の発現が上昇し,転写因子FoxM1のDNA結合活性が促進した.興味深いことに,グルコース刺激により,インスリン受容体に依存してCENP-A, PLK1の発現レベルが上昇した.これらのことから,インスリン受容体活性化は転写因子FoxM1を活性化し,標的遺伝子の発現誘導を介して細胞分裂制御に関与することが示唆された.2型糖尿病は,インスリン抵抗性の獲得と膵β細胞機能不全を特徴とする.2型糖尿病において,インスリンを分泌する膵臓β細胞の増殖促進は,膵β細胞マスの増加を促しインスリン分泌細胞を増やすことから,インスリンシグナルによる細胞分裂促進は糖尿病の進行を抑制あるいは遅延させると考えられる.
EGF受容体のリガンドであるEGF様成長因子アンフィレグリンが,EGF受容体を介し細胞分裂に関与することが報告された12).アンフィレグリンのノックダウンは細胞分裂異常を引き起こし,4n以上のDNAを持つ細胞を増加させた.このとき,多くの遺伝子の転写が変化し,それらの多くは転写因子FoxM1の認識配列を持っていた.よって,インスリン受容体同様,EGF受容体シグナルもFoxM1活性化を介し関節的に細胞分裂制御に関与することが明らかになった.EGF受容体およびその下流シグナルは細胞分裂期において阻害されていると考えられていたが,細胞分裂期においてもEGF刺激に応答してEGF受容体がリン酸化することが示されており,間期における遺伝子転写を介した関与ではなく,細胞分裂期における直接的な関与も示唆している.たとえば,EGFによるEGF受容体の活性化が,FAKキナーゼのSer732のリン酸化を亢進して細胞分裂制御に関与することが報告された13).また,EGF刺激により分裂期でEGF受容体がエンドサイトーシスされる14)ことは,細胞分裂期においてもEGFシグナルが活性化されうることを示しており,細胞分裂への関与を支持する.
本稿では,EphA2による細胞分裂制御を中心に説明した.これまで我々は,EphA2以外の受容体型チロシンキナーゼの阻害剤について細胞分裂への影響を見いだしてきた.VEGF受容体を阻害するA83-01,SU4312,Ki8751は,染色体整列異常を引き起こすことで紡錘体形成チェックポイントを活性化して細胞分裂進行を遅延させることを最近報告した15).さらに,IGF1受容体16)やALK(投稿準備中)の阻害剤が細胞分裂を遅延させることも見いだしている.IGF1受容体とALKをノックダウンしても同様な細胞分裂遅延が観察されたことから,阻害剤により観察された細胞分裂遅延は阻害剤のオフターゲット効果ではなく,これらの受容体機能を阻害したことに起因する.これらの受容体型チロシンキナーゼの細胞分裂における役割も興味深い.また,受容体型チロシンキナーゼの恒常的活性化は細胞のがん化に関与し,抗がん剤の標的となる.これらの抗がん剤の細胞増殖抑制機構の一つとして,細胞分裂への影響が考えられる.これを考慮することでより効果の高い併用療法を見いだす可能性があり,臨床への橋渡しもしたい.
1) Kaibori, Y., Saito, Y., & Nakayama, Y. (2019) EphA2 phosphorylation at Ser897 by the Cdk1/MEK/ERK/RSK pathway regulates M-phase progression via maintenance of cortical rigidity. FASEB J., 33, 5334–5349.
2) Pasquale, E.B. (2005) Eph receptor signalling casts a wide net on cell behaviour. Nat. Rev. Mol. Cell Biol., 6, 462–475.
3) Pasquale, E.B. (2010) Eph receptors and ephrins in cancer: Bidirectional signalling and beyond. Nat. Rev. Cancer, 10, 165–180.
4) Miao, H., Li, D.Q., Mukherjee, A., Guo, H., Petty, A., Cutter, J., Basilion, J.P., Sedor, J., Wu, J., Danielpour, D., et al. (2009) EphA2 mediates ligand-dependent inhibition and ligand-independent promotion of cell migration and invasion via a reciprocal regulatory loop with Akt. Cancer Cell, 16, 9–20.
5) Zhou, Y., Yamada, N., Tanaka, T., Hori, T., Yokoyama, S., Hayakawa, Y., Yano, S., Fukuoka, J., Koizumi, K., Saiki, I., et al. (2015) Crucial roles of RSK in cell motility by catalysing serine phosphorylation of EphA2. Nat. Commun., 6, 7679.
6) Hamaoka, Y., Negishi, M., & Katoh, H. (2016) EphA2 is a key effector of the MEK/ERK/RSK pathway regulating glioblastoma cell proliferation. Cell. Signal., 28, 937–945.
7) Barquilla, A., Lamberto, I., Noberini, R., Heynen-Genel, S., Brill, L.M., & Pasquale, E.B. (2016) Protein kinase A can block EphA2 receptor-mediated cell repulsion by increasing EphA2 S897 phosphorylation. Mol. Biol. Cell, 27, 2757–2770.
8) Vassilev, L.T., Tovar, C., Chen, S., Knezevic, D., Zhao, X., Sun, H., Heimbrook, D.C., & Chen, L. (2006) Selective small-molecule inhibitor reveals critical mitotic functions of human CDK1. Proc. Natl. Acad. Sci. USA, 103, 10660–10665.
9) Kawai, H., Kobayashi, M., Hiramoto-Yamaki, N., Harada, K., Negishi, M., & Katoh, H. (2013) Ephexin4-mediated promotion of cell migration and anoikis resistance is regulated by serine 897 phosphorylation of EphA2. FEBS Open Bio, 3, 78–82.
10) Dogterom, M. & Koenderink, G.H. (2019) Actin-microtubule crosstalk in cell biology. Nat. Rev. Mol. Cell Biol., 20, 38–54.
11) Shirakawa, J., Fernandez, M., Takatani, T., El Ouaamari, A., Jungtrakoon, P., Okawa, E.R., Zhang, W., Yi, P., Doria, A., & Kulkarni, R.N. (2017) Insulin signaling regulates the FoxM1/PLK1/CENP-A pathway to promote adaptive pancreatic β cell proliferation. Cell Metab., 25, 868–882.e5.
12) Stoll, S.W., Stuart, P.E., Swindell, W.R., Tsoi, L.C., Li, B., Gandarillas, A., Lambert, S., Johnston, A., Nair, R.P., & Elder, J.T. (2016) The EGF receptor ligand amphiregulin controls cell division via FoxM1. Oncogene, 35, 2075–2086.
13) Rea, K., Sensi, M., Anichini, A., Canevari, S., & Tomassetti, A. (2013) EGFR/MEK/ERK/CDK5-dependent integrin-independent FAK phosphorylated on serine 732 contributes to microtubule depolymerization and mitosis in tumor cells. Cell Death Dis., 4, 1–12.
14) Liu, L., Shi, H., Chen, X., & Wang, Z. (2011) Regulation of EGF-stimulated EGF receptor endocytosis during M phase. Traffic, 12, 201–217.
15) Okumura, D., Hagino, M., Yamagishi, A., Kaibori, Y., Munira, S., Saito, Y., & Nakayama, Y. (2018) Inhibitors of the VEGF receptor suppress HeLa S3 cell proliferation via misalignment of chromosomes and rotation of the mitotic spindle, causing a delay in M-phase progression. Int. J. Mol. Sci., 19, 4014.
16) Yamagishi, A., Ikeda, Y., Ikeuchi, M., Yuki, R., Saito, Y., & Nakayama, Y. (2020) Targeting insulin-like growth factor 1 receptor delays M-phase progression and synergizes with Aurora B inhibition to suppress cell proliferation. Int. J. Mol. Sci., 21, 1058.

京都薬科大学生化学分野教授.博士(薬学).
1967年山形県に生る.90年千葉大学薬学部卒業.92年同大学院薬学研究科博士前期課程修了.92年サンド薬品研究員,95年より千葉大学薬学部教務職員~准教授を経て,2012年現職.
研究テーマと抱負チロシンリン酸化による細胞分裂制御機構と,この制御が破綻した場合の細胞分裂への影響を解析している.新たな制御機構に関与する分子からがんの治療標的を見出し,新しい治療方法の創出につなげたい.
ウェブサイトhttp://labo.kyoto-phu.ac.jp/seika/saito/homu.html
趣味美しい景色を愛でながらのジョギング,読書.

This page was created on 2020-03-10T13:38:29.06+09:00
This page was last modified on 2020-03-30T17:38:27.000+09:00
このサイトは(株)国際文献社によって運用されています。