1962年にStanley Cohen博士によって上皮成長因子(epidermal growth factor:EGF)が発見されてから,60年を迎えようとしている1).1980年代前半までに,受容体型チロシンキナーゼ(receptor tyrosine kinase:RTK)のEGFR(EGF receptor)が同定されている2).同じころ,レトロウイルスがコードするがん遺伝子の研究から,RASなどのシグナル分子として機能するがん原遺伝子が次々発見された3).1980年代後半から1990年代前半に,マイトジェン活性化タンパク質キナーゼ(mitogen-activated protein kinase:MAPK)カスケードが発見され,受容体から転写因子につながる主要な細胞増殖のシグナル伝達経路が解明された4).また,活性化に伴うEGFRのエンドサイトーシス機構の解析は,小胞輸送研究に多大な貢献を果たしてきた.このように,EGF-EGFR経路は,細胞内情報伝達研究の先導役を担ってきた.
EGF-EGFR経路の基礎研究成果は,EGFRを標的としたがん薬物療法の開発に発展してきた.1996年にEGFRチロシンキナーゼ阻害剤(EGFR tyrosine kinase inhibitor:EGFR-TKI)のゲフィチニブが合成され,EGFの発見から40年経過した2002年に肺がん治療薬として承認された5).その後,EGFR自身の活性化変異や薬剤耐性を起こす二次変異の発見,さらには第三世代EGFR-TKIによる耐性克服など,EGFRはがん分子標的治療分野においても先導的役割を果たしている6–8).
以上のように,基礎から臨床に至るまで長い歴史を持つRTK研究ではあるが,その活性化機構についてはいまだに完全には解明されていないようである.細胞外ドメインにリガンドが結合することでチロシンキナーゼが活性化される,いわゆる定型的活性化機構は一般的に広く受け入れられている.しかし最近,リガンドが結合していないRTKが,自身のチロシンキナーゼを活性化させることもなく,細胞の中から非定型的な活性化を受ける仕組みがあることがわかってきた.特に,RTKの一つであるEphA2については,リガンド非依存的な非定型的活性化によるがん悪性化機構が注目を集めている.そこで本稿では,代表的なRTKであるEGFRとEphA2の非定型的活性化の重要性について概説し,がん分子標的治療への応用についても考えたい.
1)EGFRの定型的リン酸化
RTKはI型膜タンパク質であり,細胞内には膜近傍ドメイン,チロシンキナーゼドメインに続き,C末端側に下流にシグナルを伝える自己リン酸化部位がある.リガンドによる定型的なEGFR活性化においては,C末端領域の複数のチロシン残基が自己リン酸化される.たとえば,Tyr-1068やTyr-1086はGrb2とのドッキングサイトとなっており,下流のRAS-MAPK経路などへシグナルを伝える.また,Tyr-1045リン酸化はCblによるEGFR自身のモノユビキチン化に関わっている9).
2)EGFRの細胞内輸送
リガンドが結合した活性化EGFR二量体は,その後速やかにリガンドを結合したままエンドサイトーシスされる.このエンドサイトーシスの機構にはクラスリン依存性のものと非依存性のものがある10–12).また,EGFRのモノユビキチン化が関わっているという報告もある.リガンドとともにエンドサイトーシスされたEGFRは初期エンドソームに運ばれ,その後分解または再利用のどちらかの経路に振り分けられる.分解経路ではEGFRは多胞体(multivesicular body:MVB)とも呼ばれる後期エンドソームからリソソームに運ばれ,これによりEGFシグナルが終息する.最近,初期エンドソームからリソソームに運ばれる過程で,ROCOファミリーキナーゼLRRK1が後期エンドソームマーカーRab7をリン酸化することが報告された13).もう一方の再利用経路では,リガンドと解離したEGFRがリサイクリングエンドソームを経由して細胞膜に戻る.このような定型的活性化に伴うEGFRの細胞内輸送についてはさまざまな分子が関与することが明らかにされているが,詳細は既報の総説を参照されたい10–12).
1)EGFRの非定型的リン酸化
細胞が種々のストレスを受けた際,その応答として,EGFRの膜近傍ドメインやC末端領域にあるセリン/トレオニン残基が非定型的なリン酸化を受けることがわかっている.我々は,TNF-α(tumor necrosis factor-α)刺激によって,膜近傍領域のThr-669およびC末端領域のSer-1046/1047が,それぞれERKおよびp38によってリン酸化されることを報告した14).その後の詳細なリン酸化プロテオミクス解析により,これら以外にも数多くの非定型的なセリン/トレオニンリン酸化部位(Ser-967, Ser-1001, Ser-1013, Ser-1015, Thr-1017, Ser-1018, Ser-1021, Ser-1034, Ser-1142など)が存在していることが示されている15).後述するように,Ser-1015, Thr-1017, Ser-1018はp38によってリン酸化され,EGFRのエンドサイトーシスの引き金となることがわかっている16).
一方,膜近傍領域にあるThr-669はERKによってリン酸化され,チロシンキナーゼ活性のフィードバック阻害に関わっている17).このトレオニン残基は他のEGFRホモログ,たとえば,ErbB受容体においても高度に保存されている.EGFRはリガンドが結合すると二量体化し活性化することが知られているが,X線結晶構造解析の結果より,EGFRホモ二量体は左右非対称に形成されていることが知られている.このモデルでは,片方のキナーゼドメイン(アクチベーター側)のC-Lobeがもう一方のキナーゼドメイン(レシーバー側)のN-Lobeとドッキングして活性化するモデルが提唱されている.つまり,二つのキナーゼドメインがそれぞれ違う役割を果たしており,このうちフィードバック阻害にはレシーバー側のThr-669のリン酸化が関与している17).
2)p38に依存した非定型的エンドサイトーシス
細胞ストレスによって,p38を介してEGFRのエンドサイトーシスが誘導されることが2006年ごろから相次いで報告された.Winograd-Katzらの報告によると,EGFRを過剰発現するヒト乳がん細胞株MDA-MB-468を抗がん剤シスプラチンで処理すると,p38依存的にEGFRのリン酸化が起こった18).興味深い点は,シスプラチン刺激によってp38依存的にEGFRがエンドサイトーシスすることである.同じころ,我々を含む複数のグループが,HeLa細胞においてタンパク質合成阻害剤アニソマイシン,炎症性サイトカインTNF-αや紫外線が,同じようにp38依存性のエンドサイトーシスを誘導することを報告した19–21).また,クラスリン重鎖(clathrin heavy chain)やそのアダプター分子であるAP-2(adaptor protein-2)のノックダウン実験により,このエンドサイトーシスがクラスリン介在性であることを示した.細胞内に移行した後,EGFRはリガンド刺激時と同様に初期エンドソームに局在化するが,ストレス下ではEGFRはアクチン重合を促進するタンパク質WASHやエンドソームのソーテイングに関わるALIXなどを介してリゾビスホスファチジン酸を豊富に含む核周辺のMVBsに局在化する22).興味深いことに,TNF-αや紫外線刺激ではEGF刺激時に起こるEGFRのチロシンリン酸化やユビキチン化がまったく起こらず,そのためp38依存性エンドサイトーシスはEGFR-TKIによって抑制することができない.さらに,p38活性化がおさまると同時に脱リン酸化され,最終的に細胞膜にリサイクルされる.このp38依存的なエンドサイトーシス/リサイクル経路の発見は,以下記載のとおりリガンド刺激時のEGFRの細胞内輸送機構を再考するきっかけとなった.
3)キナーゼ活性非依存的なEGFRの機能
上述したようにある種の細胞ストレスは,EGFRの一時的なエンドサイトーシス/リサイクルを誘導する.この細胞内小胞輸送はリガンドおよびチロシンキナーゼに非依存的であり,セリン/トレオニン残基のリン酸化を介した非定型的活性化と位置づけられる23).EGFRの非定型的活性化の生理機能として,Zwangらはシスプラチンによる細胞死を抑制することから,抗がん剤耐性に関わっていることを示唆している19).また我々は,EGFRをノックダウンしたHeLa細胞は,TNF-αによるアポトーシスが起こりやすいことを示した14).このEGFRの抗アポトーシス作用は,TNF-αシグナルにおいて重要な抗アポトーシス経路であるNF-κB(nuclear factor-κB)経路とは独立しており,両者を同時にノックダウンすることで細胞はTNF-αによるアポトーシスにきわめて高い感受性を示すようになる.
リガンドやチロシンキナーゼ活性に非依存的な活性化の意義について,いくつかの興味深い報告がある.チロシンキナーゼに依存しないEGFRの機能の存在を明確に示しているのは,キナーゼ活性のないEGFRを発現するノックインマウスは眼や皮膚に異常はあるものの,EGFR欠損マウスと違って生存できる事実である24).ストレス応答の観点からは,EGFRは血清飢餓によるオートファジー誘導に関わっていることが報告されている25).血清飢餓条件において,LAPTM4B(endosomal protein lysosomal-associated protein transmembrane 4β)と相互作用し細胞内小胞にとどまっているチロシン非リン酸化EGFRは,Beclin-1をRubicon(Run domain Beclin-1–interacting and cysteine-rich–containing protein)から遊離させ,オートファジーを誘導する.一方,EGF存在下では,チロシンリン酸化EGFRはBeclin-1のチロシンリン酸化を介してオートファジーを抑制する26).興味深いことに,血清飢餓ストレスを含めた多くのオートファジー誘導条件においては,p38の活性化が起こることが知られている.したがって,p38による非定型的なEGFRリン酸化がオートファジーの制御に関わっている可能性が考えられる.
リガンド誘導性の定型的EGFR活性化に伴う細胞内輸送については数多くの報告がある.一方,エンドサイトーシスした後,EGFRがどのようにしてリソソーム経路とリサイクル経路へ振り分けられるのかという問題は長い間解決できていない大きな課題として残されていた.これに関連して,高濃度リガンド刺激ではほとんどのEGFRがリソソーム分解を受けるのに対して,低濃度では主に細胞膜へリサイクルされるという現象も観察されていた.しかし,従来モデルではリガンド濃度が変化しても,エンドサイトーシスするEGFR分子にEGFが結合していることに変わりはなく,エンドサイトーシス後になぜ異なる経路に振り分けるのかは不明なままであった.
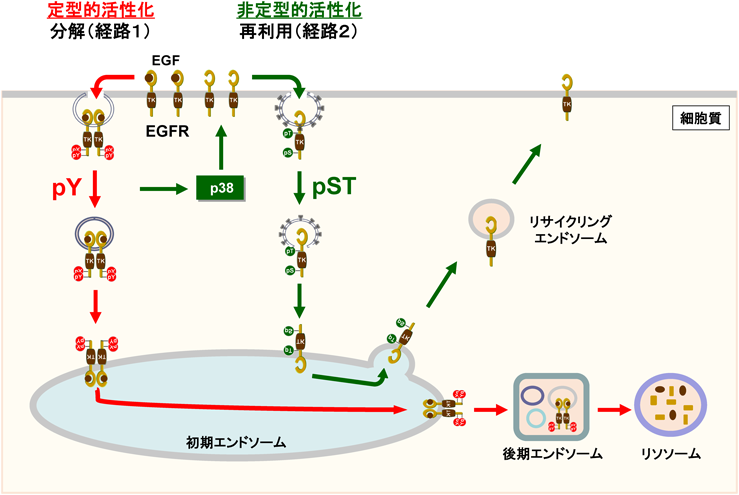
そこで我々は,この振り分け機構を説明できるデュアル輸送モデルを提唱した(図1)16).EGFによるEGFR活性化がストレス応答と同様にp38を活性化することから,リガンド刺激時にもp38依存的なエンドサイトーシスが起こると考えた.つまりこのモデルは,従来モデルで考えられていたEGFが結合したEGFR二量体の定型的なエンドサイトーシス(経路1)に加えて,単量体EGFRのp38依存的な非定型的エンドサイトーシス(経路2)が同時並行で進行しているというものである.また,ストレス応答でp38依存的にエンドサイトーシスしたEGFRは細胞膜にリサイクルされることから,経路1でエンドサイトーシスしたEGFRがリソソーム分解経路に,経路2によるものがリサイクル経路にそのまま運ばれると考えた.つまり,このモデルではそもそもエンドサイトーシス機構が二つあり,どちらで細胞内移行するかによってその後の輸送経路が運命づけされていることになる.また,このモデルでは,リガンド濃度によって輸送機構が異なる現象にも説明がつく.リガンド濃度が高い場合(100 ng/mL),刺激直後に多くの受容体にリガンドが結合するため,エンドサイトーシスするEGFRはほとんど定型的な経路1に運ばれる.一方,リガンド濃度が低い場合(3 ng/mL),リガンドが結合した一部のEGFRを除き,細胞表面上の多くのEGFRはリガンドが結合しない単量体のままであり,それらは細胞の中からp38によってリン酸化されることで非定型的な経路2に運ばれることになる.
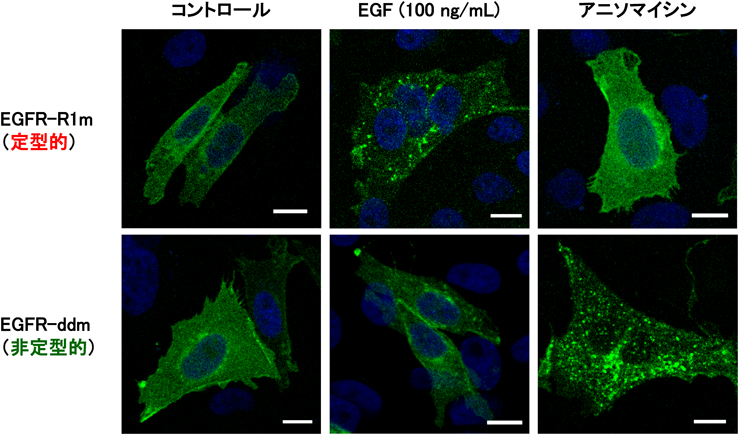
以上のように,デュアル輸送モデルはこれまでのEGFR研究の課題を解決するユニークなモデルである.すでに我々は,このモデルを実証する論文を公表している16).その中では,非定型的なエンドサイトーシスにはp38によるSer-1015/Thr-1017/Ser-1018のリン酸化が必要であること,定型的なエンドサイトーシスにはEGFRの二量体化によるチロシンキナーゼ活性化が必須であることを示している.図2には,定型的なエンドサイトーシスのみが認められるEGFR変異体(R1m:Ser-1015, Thr-1017, Ser-1018をアラニンに置換)と非定型的な活性化のみ認められる変異体(ddm:二量体化に必要な細胞外および細胞内のドッキング部位を変異させたもの)をCHO-K1細胞に発現させたときの結果を示す.高濃度EGFで刺激すると,R1mのエンドサイトーシスは起こっているが,ddmのエンドサイトーシスは認められない.逆に,p38を活性化するアニソマイシンで刺激した場合は,R1mのエンドサイトーシスは起こらないが,ddmのエンドサイトーシスは認められている.このように,これらEGFR変異体は,エンドサイトーシス機構を解析する有用なツールとして利用することができる.さらに,細胞膜にリサイクルしてきたEGFRはEGFと結合可能であることも示しており,リサイクリングによって受容体の再利用が行われていることも明らかにしている.このワーキングモデルで重要なことは,受容体がそのリガンドによって活性化される場合,リガンドが結合したものと結合しなかったものがそれぞれ別々の機能を果たしているということである.今後,他のリガンド-受容体間でも同じような現象が認められるのかについても解析が必要である.
1)EphA2の定型的活性化
Eph受容体ファミリーは14種あり,エリスロポエチンを産生する肝細胞がんに発現する受容体型チロシンキナーゼとして同定されたことから,EphA1(erythropoietin-producing hepatocellular receptor A1)と名づけられた27).Eph受容体の中で最も研究が進んでいるのは1990年に同定されたEphA2であり,血管新生や発生のプロセスにおいて重要な役割を果たしている28–30).GPIアンカー型の膜結合型リガンドEphrin-A1が隣接する細胞に発現するEphA2と結合すると,チロシンキナーゼの活性化により下流にフォワードシグナルを伝える.一方,リガンドが発現する細胞側にも,リバースシグナルが伝わる.リガンドによるEphA2活性化はFAK, AktやERKを抑制し,細胞増殖や遊走が負に制御される.したがって,定型的なEphA2活性化は上皮性の維持に重要であり,がん抑制シグナルとして働いている31, 32).
2)EphA2の非定型的リン酸化
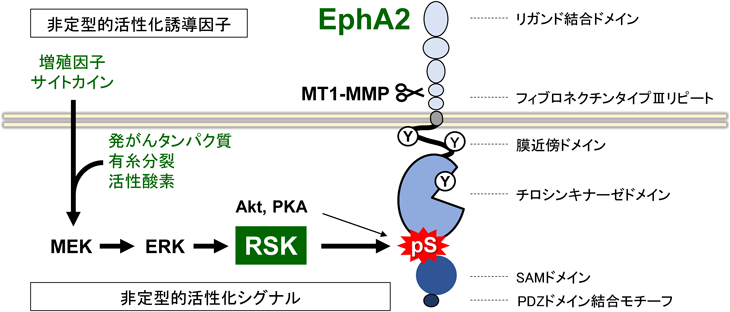
EphA2の過剰発現が多くのがん組織で観察され,その発現レベルはがん悪性度と正の相関を示す32–34).しかし,そのリガンドの発現はしばしば抑制されていることから,EphA2のリガンドやチロシンキナーゼ活性に依存しない非定型的活性化の存在が示唆されてきた.実際,EphA2にも多くのセリン/トレオニンリン酸化部位が見つかっており,キナーゼドメインとSAMドメインの間のリンカー部位には5個のリン酸化部位が同定されている(図3)35).
2009年,Miaoらは神経膠芽腫細胞を血清刺激することでAktを介してSer-897リン酸化が誘導されることを示した36).しかし我々は,神経膠芽腫細胞をはじめとするさまざまながん細胞において,Ser-897リン酸化がAktではなく,ERKの下流キナーゼであるリボソームS6キナーゼ(p90 ribosomal S6 kinase:RSK)に依存することを示した37).RSKによるリン酸化は血清刺激だけでなく,EGFなどの成長因子やTNF-αなどの炎症性サイトカインでも誘導され,in vitroキナーゼアッセイにより,RSKがEphA2 Ser-897リン酸化を触媒することを明らかにした.一方,前立腺がん細胞では,PKAがSer-897とともに,Ser-892, Thr-898, Ser-899, Ser-901のリン酸化を誘導すると報告されている38).これらSer-897をリン酸化する3つのキナーゼはいずれもAGCファミリーに属しており,その基質コンセンサス配列はよく似ているものの,我々はEphA2 Ser-897の非定型的リン酸化は主にRSKが担っていると考えている.
一方,神経膠芽腫細胞にEphA2を過剰発現させると,EphA2チロシンキナーゼの活性化がERK-RSK経路を活性化し,Ser-897リン酸化を誘導する39).また,EphA2が膜結合型マトリックスメタロプロテアーゼMT1-MMPによってN末端側のフィブロネクチンタイプIIIリピートで切断され,リガンド結合ドメインを持たない切断型EphA2が生じ,この変異体もSer-897がリン酸化されることが示されている40, 41).以上のことから,EphA2の非定型的リン酸化はリガンドの有無にかかわらず,様々な条件で誘導される.
3)非定型的活性型EphA2の生理機能
EphA2は上皮性維持に関わることが知られている.イヌ腎臓細胞MDCKを肝細胞増殖因子(hepatocyte growth factor:HGF)存在下で3D培養すると,中心の管腔を取り囲むように極性を持つ細胞が単層を形成した袋状の嚢胞構造をとる.管腔形成の最初のステップとして上皮間葉転換(epithelial-to-mesenchymal transition:EMT)が部分的に誘導され,基底膜側から外側へアクチンが豊富な突起を伸張させる.このとき,EphA2は基底膜に主に局在化し,Ser-897リン酸化が嚢胞構造の伸張に関与している42).さらに嚢胞構造の伸張にはRhoファミリーのグアニンヌクレオチド交換因子(guanine nucleotide exchange factor:GEF)であるEphexin4とEphA2の結合,それに伴うRhoGの活性化が必須である.
EphA2は有糸分裂にも関わっている43).G2期においては定型的なTyr-588リン酸化がみられるのに対して,M期中期にTyr-588が完全に脱リン酸化されており,代わりに細胞増殖シグナルがCDK1(cyclin-dependent kinase 1)を介してMEK-ERK-RSKを活性化することでSer-897リン酸化が誘導される.そこへEphexin4が結合し,RhoGの活性化が誘導されることでM期が正常に進行する.このように,細胞周期の進行に伴うEphA2の定型的活性化から非定型的活性化へ急激な変化は,非定型的活性化の重要性を如実に示す事例として注目される.
1)腫瘍組織における非定型的活性型EphA2の発現上昇
EphA2は肺がん,胃がん,大腸がん,膵臓がん,乳がん,神経膠芽腫,悪性黒色腫などにおいて過剰発現が認められ,がんのステージや患者予後に相関する.神経膠芽腫では,Ser-897リン酸化EphA2がグレードIV,特に浸潤性の細胞において発現が著しく高い36).上咽頭がんにおいては,リン酸化EphA2が原発巣だけでなく,転移リンパ節においても認められる44).浸潤性乳がんやユーイング肉腫では,リン酸化EphA2が患者予後と負の相関を示す45, 46).また,細胞外ドメインが切断された非定型的活性型EphA2は様々な腫瘍組織でMT1-MMPとの共発現が認められ,浸潤性乳がんのリンパ節の転移巣でもその共局在が認められる40, 41, 47, 48).切断されたEphA2細胞外ドメインは患者血中に検出されることから,新たな診断マーカーとしても期待される49).
我々は,腫瘍組織における非定型的EphA2活性化におけるRSKの重要性について調べた37).組織マイクロアレイを用いた結果,肺がん,乳がん,甲状腺がん,肝がん,大腸がん,前立腺がん,卵巣がん,子宮体がん,腎細胞がん,膵臓がん,胆管がん,尿道上皮がんにおいて,活性型RSKとSer-897リン酸化EphA2の共局在が認められ,EGFR活性化変異を持つ肺腺がん患者検体においても,両者の共局在が認められた.また,活性型RSKと非定型的活性型EphA2が共発現している肺がん患者は,著しく予後が悪いことがわかった.
以上の報告をまとめると,EphA2のSer-897リン酸化は悪性度の高い腫瘍において誘導されており,特に転移性腫瘍での発現も高い.したがって,EphA2の非定型的活性化はさまざまながん腫において,新しい治療ターゲットとなることが期待される.
2)非定型的EphA2活性化によるがん細胞の増殖制御
非定型的活性型EphA2は,がん細胞の増殖にも関与する.胆管腺がん細胞において,EphA2の非定型的活性化によってAkt-mTORC1経路,RAF-MEK-ERK経路,Pyk2-Src-ERK経路が亢進している50).低分化型胆管腺がん細胞ではEphA2リガンドの発現低下とSer-897リン酸化が起こり,中分化型胆管腺がん細胞にEphA2を導入すると軟寒天培地におけるコロニー形成やマウス異種移植モデルにおける腫瘍形成の促進が認められている.神経膠芽腫細胞では,EGF誘導性の細胞増殖にRSK-EphA2経路が必須である51).さらに,上咽頭がん,胃がん,ユーイング肉腫においても同様の報告があり,MT1-MMPによる切断型EphA2も腫瘍形成に関わることが報告されている41, 44, 46).
EphA2の非定型的活性化が,足場を確保できない細胞がアポトーシスを起こす現象であるアノイキスへの耐性化に関わることが示されている52).また,がん抑制因子としての働くEphB6がEphA2と二量体を形成し,Ser-897リン酸化を抑制することで乳がん細胞のアノイキス耐性を解除することも知られている53).神経膠芽腫では,グルコース枯渇時の活性酸素産生にシステイン/グルタミン酸トランスポーターxCTが関与するが,このときERK-RSKによるEphA2リン酸化が細胞生存シグナルとして働いている54).
3)細胞運動におけるEphA2の非定型的活性化
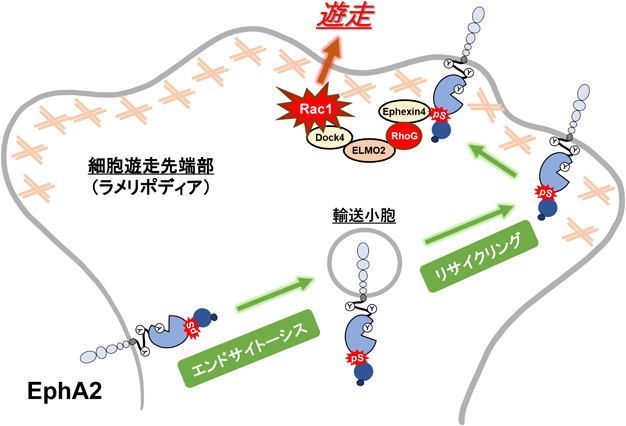
EphA2の非定型的活性化の最も重要な機能は,細胞運動能の制御である(図4).神経膠芽腫,乳がん,肺がん細胞において,EphA2 Ser-897リン酸化が細胞遊走を促進することが示されている36, 37).我々は,運動能が高い乳がん細胞MDA-MB-231において,Ser-897リン酸化EphA2が過剰発現し,しかも細胞の遊走先端に局在化することで細胞運動能を制御することを報告した37).さらに,細胞–細胞間の反発作用の制御にも,EphA2のSer-897リン酸化が関わっている55).HGFによってEphA2の非定型的活性化が起こる際,LMTK3(Lemur tyrosine kinase-3)によってリン酸化されたRCP(Rab-coupling protein)は,小胞輸送におけるリサイクリング調節因子Rab14と結合することでEphA2を細胞膜へ輸送する.その結果,細胞どうしが反発し合い,細胞の遊走や浸潤を誘導する.前立腺がん細胞においては,神経軸索ガイダンスの反発因子として有名なEphA2リガンドEphrin-A1により細胞周囲の突起構造が退縮するが,これはPKAによるEphA2 Ser-897リン酸化で回避できる38).つまり,Ephrin-A1による定型的EphA2活性化とSer-897リン酸化による非定型的活性化が相反する作用を示し,両者の調節が細胞遊走能の制御に重要である.
非定型的活性型EphA2による細胞運動能亢進機構の解析により,EphA2の非定型的活性化が誘導されると,EMT誘導やM期進行のときと同様にEphexin4と結合し,RhoGを活性化する52).RhoGの下流のエフェクター分子ELMO2とRacのGEFであるDock4の複合体が細胞膜へ運ばれ,EphA2-Ephexin4-ELMO2-Dock4の複合体を形成する.その結果,Dock4によるRac1の活性化を誘導し,アクチン骨格の再構成によって細胞運動が調節されている.
MT1-MMPによる切断型EphA2も細胞運動を制御する.野生型,もしくは切断型EphA2を発現した乳がん細胞をSCIDマウス乳腺に同所性に移植すると,切断型導入細胞では細胞間の結合能が減弱し,間質への浸潤が認められる40).切断型導入細胞ではリンパ節転移も認め,転移した細胞は細胞間接着が非常に弱い.また,類表皮がん細胞の実験的肺転移モデルにおいて,切断されない変異体発現細胞では肺転移が有意に低下した41).
4)非定型的活性型EphA2を介したがん幹細胞性の維持と抗がん剤耐性化
神経膠芽腫は自己複製能と未分化能を有しており,がん幹細胞としての性質を持つことが知られている.EphA2は神経膠芽腫細胞において発現が高く,EphA2発現抑制はがん幹細胞性の低下につながる56).EphA2の非定型的活性化は,膠芽腫幹細胞の自己複製能や浸潤能,ニューロスフェア形成や幹細胞マーカーの発現亢進に関与している57).また,上咽頭がん,悪性黒色腫や肺がんにおいても,EphA2はスフェア形成を誘導し,幹細胞マーカーの発現と相関を示す44, 58, 59).これらの報告から,非定型的活性型EphA2はがん幹細胞性の誘導・維持にも寄与することが示されている.
我々は,BRAF活性化変異,EGFR活性化変異,EML4-ALK融合タンパク質をそれぞれ有する悪性黒色腫細胞株,肺がん細胞株において認められる非定型的EphA2活性化が,それぞれの分子標的薬により抑制されることを示した37).しかし,分子標的薬に耐性を示す細胞において,しばしばEphA2の過剰発現が認められる.たとえば,BRAF阻害剤ベムラフェニブに耐性化した悪性黒色腫細胞やEGFR阻害剤ゲフィチニブに耐性化した肺腺がん細胞において,EphA2の発現上昇と非定型的活性化が認められる60, 61).実際,臨床においてもEGFR活性化変異を持つ肺腺がん患者において,EGFR阻害剤エルロチニブ投与前と再発後の組織切片を抗EphA2抗体で免疫染色すると,再発後にEphA2の発現上昇が認められている62).さらに,放射線照射によってRSK-EphA2経路の活性化が誘導されるが,これは放射線治療への抵抗性に寄与することが示された63).これらの報告から,EphA2は化学療法や放射線療法に対する耐性化に寄与しており,その阻害剤の開発は耐性化の克服にもつながると期待される.
EGFRやEphA2以外のRTKにおいても,セリン/トレオニンのリン酸化の役割を解析している例がある.線維芽細胞増殖因子受容体(fibroblast growth factor receptor 1:FGFR1)のC末端領域にあるSer-777がp38を介してリン酸化され,それが細胞外にあるリガンドFGF1の細胞質内や核内への移行に必須であるという報告がある64).アニソマイシンや高浸透圧ストレスによって促進されることから,EGFRと同様にp38によるFGFR1のリン酸化が,リガンド/受容体の細胞内局在を制御している可能性も考えられる.別の報告では,Ser-777はERKによってリン酸化され,FGFR1のネガティブフィードバック機構として働き,この阻害機構を回避することで細胞増殖,遊走,軸索伸長が増強することが示されている65).さらに,FGFR1はRSK2によってSer-789がリン酸化されることで,細胞内にエンドサイトーシスすることが示されている.このリン酸化を起こさなくするとFGFR1のシグナルが持続することから,このエンドサイトーシスはEGFRの定型的活性化経路と同様に,FGF1によるFGFR1シグナルを終息させるメカニズムであると考えられる66).
FGFR2においても,FGFR1のSer-777に相当するSer-780のリン酸化がFGFR2のフィードバック阻害に関与していることが報告されている.この部位を変異させると,FGFR2の活性化の持続が認められている.興味深いことに,Ser-780がロイシンに置換したS780L変異体が膀胱がんで見つかっている.この部位の変異は,FGFR2の活性を上昇させ,遊走・浸潤能が亢進していることが示されている67).
また,血管内皮細胞増殖因子受容体(vascular endothelial growth factor receptor 2:VEGFR-2)のセリン残基のリン酸化についての報告もある68).C末端領域のPEST配列の中のSer-1188とSer-1191のリン酸化がE3ユビキチンリガーゼとの結合を誘導し,VEGFR-2のプロテオソーム分解を起こすことが示されている.しかし,これらセリン残基のリン酸化を触媒するキナーゼ等はわかっておらず,今後の解析が必要である.