高血圧は日本人の成人の約4000万人が罹患しており,児童においても年々増加傾向がみられる.高血圧は心血管病の最大の危険因子であり,毎年約10万人が高血圧を原因とする脳卒中や循環器病によって亡くなっていると推定される.
高血圧は食塩の過剰摂取,肥満,精神的ストレス,睡眠時無呼吸等,さまざまな要因で起こることがわかっている.中でも食塩の過剰摂取(日本人では~10 g/日)はよく指摘されるところであり,1日に1 g以下の塩分しか摂取しない南米のヤノマミインディアンは生涯にわたって高血圧を示さないことから,減塩の重要性には疑う余地がない.
食塩の過剰摂取だけでなくさまざまな要因で起こる高血圧を合理的に説明するため,最近10年あまりの間に,中枢の交感神経制御中枢の恒常的活性化による末梢血管系や心臓・腎臓機能の変調がその根底にあると考えられ始めた(神経性高血圧).本稿では,交感神経系の恒常的活性化をもたらす原因とそのメカニズムについて最近の知見を概説する.
食塩の過剰摂取によってなぜ高血圧が生じるかという機序については,長い間,血液中のナトリウムイオン(Na+)濃度が上昇するために浸透圧差によって血管外から水が流入し,血液量が増大するためであると説明されてきた.食事に含まれる塩分はまず血液中に取り込まれるが,速やかに体液(血液や脳脊髄液をはじめとする細胞外液の総称)全体に移行するため,血液中のNa+濃度だけが慢性的に高くなるとは考えにくい.また,腎臓機能が正常であれば過剰なNaは尿中に排泄されることから,慢性的な高血圧を説明する機序としては,説得力に欠けるように思われる.一方で,高血圧は腎臓でのNa排泄にとっては有利であり(圧利尿),高血圧は生体の適応現象ともいえる.
高食塩食によって血圧上昇がみられる“食塩感受性”を示す集団は正常血圧のヒトの約30%にすぎない.通常使われる実験動物においても,水を自由に摂らせるかぎり,高食塩含有餌によって高血圧を示すことはない(後述参照)1, 2).一方,高血圧を発症しているヒトの大部分は食塩感受性を示す1, 2).そこで,Dahl-salt-sensitiveラット(DSR)3),自然発症高血圧ラット(SHR)4)やdeoxycorticosteroneモデル(DOCA-saltモデル)5)等の動物モデルが食塩による高血圧発症機構の研究対象とされてきた.
1)脳脊髄液中のNa+濃度
ラットに対して離乳期から8週間にわたって高食塩食(2%含有)を与えても高血圧を示さなかった6).ところが最近になり,12週間にわたって与え続けると高血圧を発症することがわかった7).この血圧上昇は神経節ブロックによって大きく低下することから交感神経性の血圧上昇である.おもしろいことに,このとき,血中のNa+濃度は対照と同じく生理的レベルであるのに対し,脳脊髄液中のNa+濃度が有意に高くなっていた7).これは,高血圧を示すDSRやSHRにおいて,血中ではなく脳脊髄液中のNa+濃度上昇が,血圧上昇に先行して起こるという知見と一致する8).
2)感作/神経可塑性
遺伝的素因に加えて,幼若期(発達期)に経験した環境要因がエピジェネティックな変化を通して脳・神経系の機能にも影響を及ぼすということがわかってきた.たとえば幼若期の栄養不足は,成人後のさまざまな慢性病のリスク要因となるというDOHaD説(developmental origins of health and disease)が提唱されている9, 10).
幼若期だけでなく,成体においても過去の病因への曝露経験が,その後の脳・神経系のさまざまな刺激に対する反応性に影響を与えることが知られている.オスラットに14~17日間高食塩餌を与えると,そのNaCl量に応じて,後の脳室内へのNaCl溶液の注入による交感神経の活性化と血圧上昇反応に増強効果がみられる11).ラットの脳室内に1週間にわたって血圧に影響しない量のアンジオテンシンII(AngII)を注入し,その後1週空けて,3~4週間にわたってもう一度AngIIを注入すると,AngIIに対して増強された反応が現われる12).このAngII誘導性の高血圧発症の感作は,事前のアルドステロン13),高脂肪食14),レプチン15)やTNF-α14)等への曝露によっても生じる.また,血圧に影響を与えない量のAngIIあるいはアルドステロンによる前処置の効果は,2%食塩水負荷による高血圧誘導に対してもみられる16).この中枢感作には,レニン-アンジオテンシン-アルドステロン系(RAAS),NMDA受容体,BDNFによるニューロン作用,および転写調節因子の関与などが推定されている17).
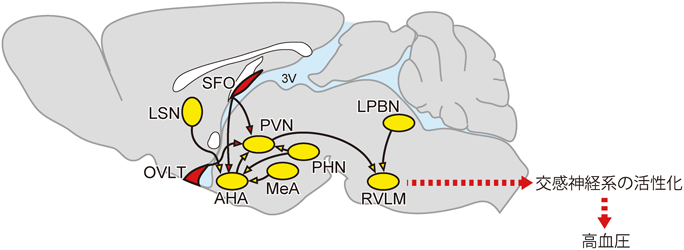
体液中のNa+濃度の上昇は脳内で感知され,その情報は交感神経制御中枢に伝えられると推定されていたが,それがどこでどのように行われているのかについては未解明のままであった.我々は,細胞外液のNa+濃度が生理的濃度である~145 mMを超えると開口を始めるNa+濃度感受性チャンネル,Naxを同定し,これまでにこれが脳内Na+濃度センサーであることを証明してきた18–40).Naxは,脳内で例外的に血液-脳関門(BBB)を欠く脳室周囲器官(CVOs)に属する脳弓下器官(subfornical organ:SFO)や終板脈管器官(organum vasculosum lamina terminalis:OVLT)の特殊なグリア細胞[アストロサイトと上衣細胞(タニサイト)]に発現している25).CVOsの中でもSFOとOVLTは器官内に神経細胞が存在していることから,最後野(AP)と併せて感覚性(sensory)CVOsと呼ばれる.NaxはSFOやOVLTにおいて体液中のNa+濃度の上昇を感知し,その情報は体液状態に応じた水分や塩分の摂取行動の制御に使われる.すなわち,SFOにおけるNaxのシグナルは塩分の摂取抑制に18, 20, 22, 29, 38),OVLTにおけるNaxのシグナルは水分の摂取誘導に利用されている35, 38, 40).
1)Nax欠損マウスは体液中のNa+濃度上昇に伴う血圧上昇を示さない
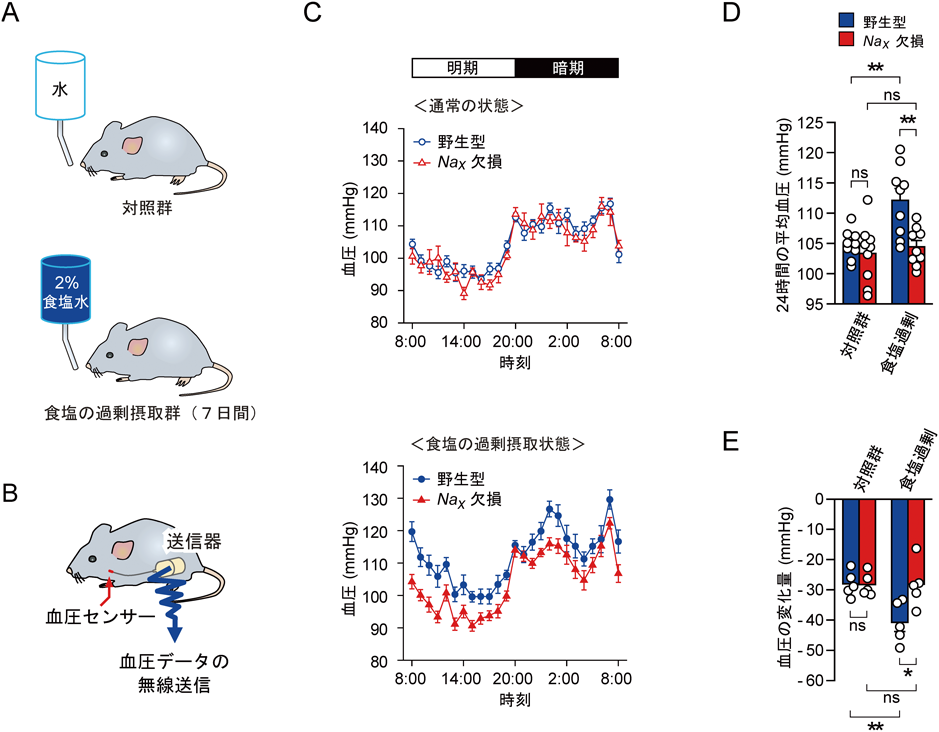
短期間で体液中のNa+濃度を上昇させる実験では,食塩含有餌に加えて,水に代えて高濃度食塩水を飲ませる方法が採用されてきた.野生型マウスとNax欠損マウス(Nax-KO)に対して,水に代えて2%の食塩水を1週間にわたって与えると(図1A, B),血液と脳脊髄液中のNa+濃度はそろって約10 mM上昇する41).この時,野生型マウスでは平均血圧が有意に上昇するが(~7 mmHg),Nax-KOマウスでは血圧の上昇はまったくみられない(図1C, D)41).
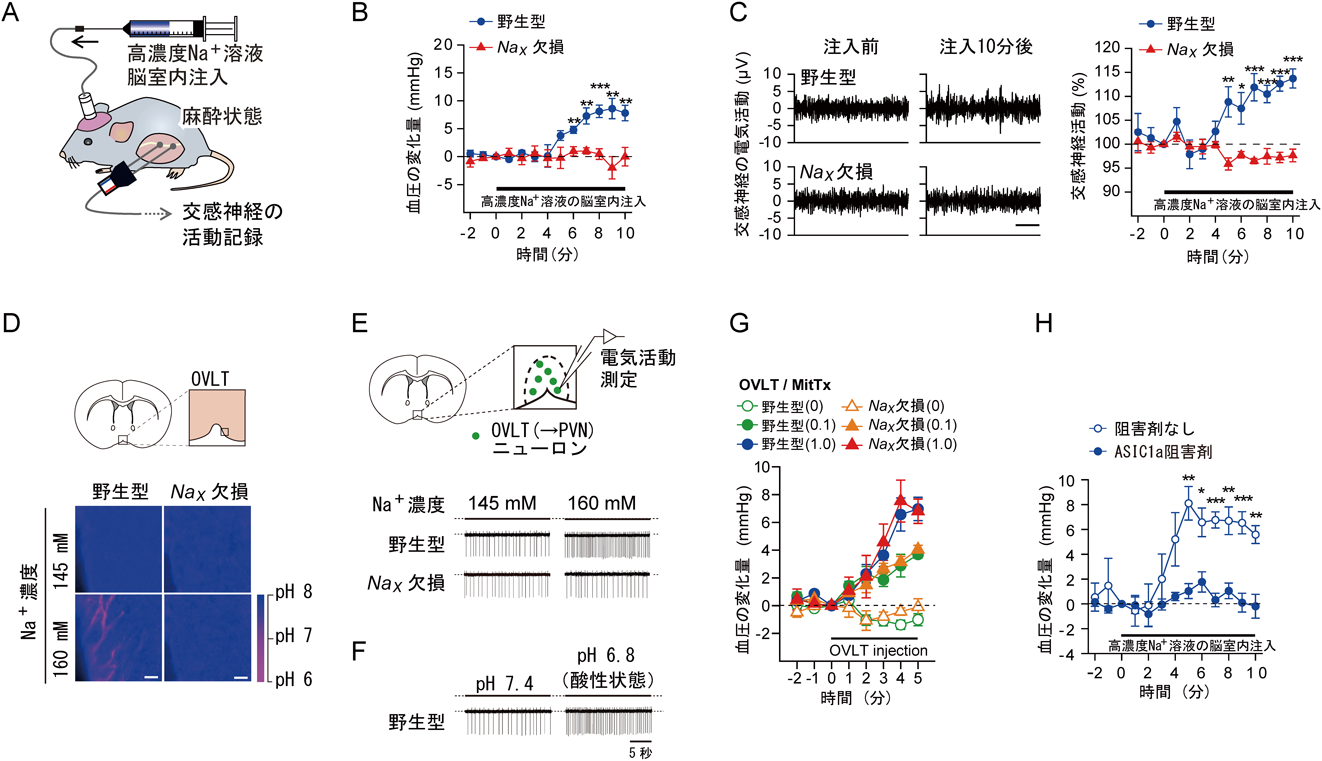
自律神経節ブロッカーによって,野生型マウスの方に,より大きな血圧降下がみられることがわかる(図1E;~41 vs ~28 mmHg).高濃度Na+溶液を脳室内に投与しても,Nax-KOマウスでは血圧上昇が誘導されない(図2A, B)41).これはNax-KOマウスでは体液中のNa+濃度の上昇を感知できないためであると推定された.このとき,腰部交感神経活性をみても,野生型マウスのみに活性の増大が認められる(図2C)41).OVLTを選択的に欠失させた野生型マウスではこれらの血圧上昇が消失したことから,OVLTにおけるNaxによる体液Na+濃度上昇の感知が血圧上昇の初発段階であると考えられる41).
2)NaxシグナルはOVLT内でH+を介してニューロンに伝えられる
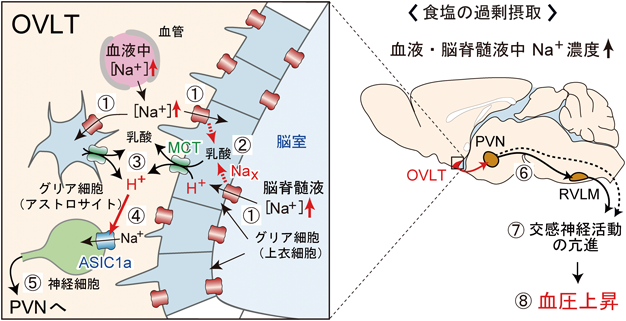
それではグリア細胞が感知したNa+濃度上昇の情報はどのようにして,ニューロンに伝達されているのだろうか? Nax陽性のグリア細胞では,細胞外Na+濃度の上昇に反応して,糖の取り込みとその嫌気的代謝活性が亢進することがわかっている26).嫌気的解糖系の最終産物である乳酸は,H+とともに乳酸/H+共輸送体によって細胞外へ放出される(図2D).OVLT中のニューロンには酸感受性チャンネルASIC1aを発現している興奮性ニューロンが存在する41).ASIC1aの活性化剤をOVLTに注入すると血圧が上昇する(図2G)が,ASIC1aの阻害剤存在下では高濃度Na+溶液を脳室内に注入しても血圧は上昇しない(図2H)41).この血圧上昇は交感神経活動の阻害剤を投与しておくと起こらないことから,交感神経を介したものである41).このようにNaxのシグナルはH+によってニューロンに伝えられているが(後述参照),同時に分泌される乳酸もASIC1の活性化を助けていることがわかった(図3,左)41).
3)NaxシグナルはOVLTからPVNに送られる
Na+濃度上昇のシグナルは,交感神経制御中枢である室傍核(paraventricular nucleus:PVN)や頭側延髄腹外側野(rostral ventrolateral medulla:RVLM)の活性化を通して,末梢の交感神経の活性化につながっていると推定されている.スライスパッチ法で細胞外Na+濃度を160 mMに上げると野生型マウスではOVLT(→PVN)ニューロン活動の活性化がみられるのに対し,Nax欠損マウスでは変化がみられない(図2E).OVLT(→PVN)ニューロンに選択的に光感受性陽イオンチャンネルChR2を発現させ光活性化すると,光強度依存的に血圧が上昇する41).この上昇は交感神経活動の阻害剤により消失する41).野生型マウスのOVLT(→PVN)ニューロンは細胞外のpHの低下に応答して活性化する(図2F)41).AS1C1aを阻害すると,Na+濃度の上昇に依存したOVLT(→PVN)ニューロンの活性化とともに交感神経性の血圧上昇も消失する(図3,左)41).Nax-KOマウスでは食塩負荷によるPVNやRVLMにおけるFosの発現上昇がみられないことから,NaxシグナルはOVLT(→PVN)ニューロンからPVNを経て,下流の交感神経制御中枢であるRVLMへと伝えられている(図3,右)41).
1)肥満と交感神経活性
肥満に伴う高血圧の発症に交感神経系が関わることはよく知られている42).メスのラットを15日間高カロリー食で飼育するだけで腹部交感神経活動の上昇がみられる43).ウサギにおいても高カロリー食によって腎臓交感神経の活性化が認められる44).
肥満における交感神経の活性化はレプチンやインスリンを含む多くの因子の作用によると考えられている42).レプチンやインスリンの遮断薬を脳室内に投与すると,高脂肪食によって肥満したウサギの高血圧を低下させることができる45).レプチンやインスリンの交感神経中枢への作用は弓状核(arcuate nucleus:ARC)を介していると考えられている46, 47).ARCからレプチン受容体を欠失させた肥満マウスでは,レプチン誘導性の腎臓交感神経の活性化と血圧上昇はみられない48).レプチンのこの作用は,POMCニューロンのメラノコルチン経路を通して行われている48–50).
この経路と並行するニューロペプチドY(NPY)/AgRPニューロンによるNPY経路も交感神経活性の制御に関与している.ARCのNPYニューロンを活性化すると,内臓交感神経活性を持続的に抑制することができる51).また,PVNへNPYを注入すると用量依存的に交感神経活性を抑制する52)とともに,ARCを介したインスリンによる交感神経の活性化を抑制する53).
レプチンは,ARC以外にも視床下部の腹内側核(ventromedial hypothalamic nucleus:VMH)54)や脊内側核(dorsomedial hypothalamus:DMH)55),またSFO56)を通して,交感神経を活性化しているという報告がある.この内,VMHやDMHは血液-脳関門の内側にあり,通常では血中のレプチンが到達しているとは考えにくい.ただし,脳内に炎症が生じている場合には脳内の血管透過性が亢進していることから,レプチンの到達もありうるかもしれない.一方で,ヒトの肥満に伴う高血圧には,レプチンは関与していないという指摘もある57).
2)肥満と炎症
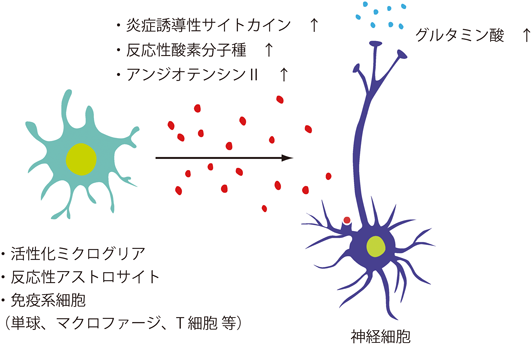
肥満は軽度ながら慢性的な炎症をひき起こすことが知られている58).脂肪組織はトリグリセリドの形でエネルギーを貯蔵する器官であると同時に,内分泌器官として重要な働きをしており,アディポサイトカインと呼ばれる,IL-1, IL-6, IL-8, IL-12, IFN-δ, TNF-α, TGF-β, LIF, MCP-1, MIP-1,レプチン等の一群の分子を生成する59).これらの分子の生成は,脂肪組織において細胞死を誘導し慢性的な炎症を起こす60)と同時に,脳・神経系をはじめとして他のシステムにも影響を与え,その機能に異常を生じさせる61).脂肪組織においてはM1マクロファージが活性化するが,同時に脳ではミクログリア細胞の活性化が起こる.これがSFO, PVN, RVLM等の慢性的炎症と神経活動の亢進を促し,交感神経系の恒常的な活性化および血圧の上昇につながっていると考えられている(下記参照).
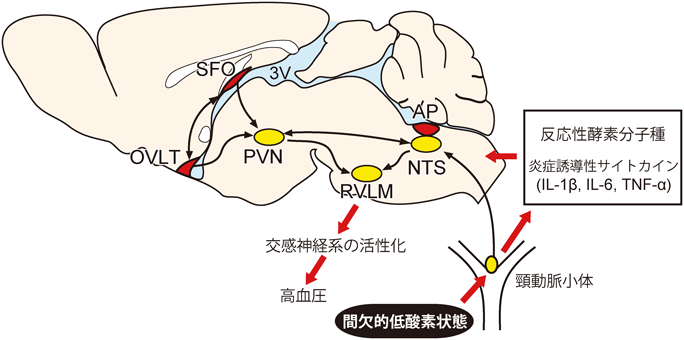
睡眠時無呼吸症候群のような慢性的に間欠的な低酸素曝露が生じる場合にも高血圧が発症する62).本症候群では,血液中の酸素濃度の低下を検知する頸動脈小体(carotid body:CB)に異常な興奮が認められる63).酸素濃度の低下/再上昇の繰り返しに伴い,反応性酸素分子種(ROS)や反応性窒素分子種(RNS)が生成し,化学受容器をもつ錐体神経に異常な発火が生じると推定されている64, 65).同時に,血中のTNF-α, 1L-1β, 1L-6, C-reactive protein等の炎症性サイトカインの上昇や66, 67),孤束核(nucleus tractus solitarius:NTS)におけるジャンクション接着分子(JAM-1)の発現上昇がみられる(図4)68).
錐体細胞はNTSに投射しており69, 70),NTSの興奮性ニューロンはPVN, RVLM, SFO等に連絡している71–73)(図4参照).これらの神経核においても活動の慢性的亢進がみられることから,酸化ストレス,炎症,交感神経系の活性化が高血圧発症のメカニズムと考えられる(図4)65, 74, 75).
精神的ストレスが高血圧症の原因となることは疫学的調査から示されている76).その経路については内側扁桃体(medial amygdala:MeA)の役割が言われているが77),十分には解明されていない.急性のストレス反応として,交感神経-副腎髄質系のカテコールアミンと,視床下部-下垂体-副腎皮質(HPA)系のコルチゾール78, 79),アルドステロン80)が知られている.HPA系のCRH-ACTH-コルチゾールのカスケードでは,コルチコトロピン分泌ホルモン(CRF)が,下垂体前葉からのACTHの分泌を促し,ACTHは副腎皮質に作用してコルチゾールとアルドステロンの分泌を促す.
レニン-アンジオテンシン-アルドステロン系(RAAS)は精神的ストレスと密接に関係しており,ストレス下では血中のレニンとAngIIのレベルが上昇する81, 82).薬理学的にRAASをブロックすると気分が改善し,不安が減弱することが報告されている83, 84).
1)アルドステロン
グルココルチコイド(コルチゾール)とミネラルコルチコイドを結合する二つの受容体が知られている:タイプIグルココルチコイド受容体(MR)とタイプIIグルココルチコイド受容体(GR)である.コルチゾールは肝臓でのアンジオテンシノーゲンの生成を活性化することで血圧上昇に関わる78).アルドステロンは塩欠乏時に副腎皮質で生成されるホルモンで,塩分欲求を亢進するとともに,腎臓においてNaの再吸収を促進するホルモンとして知られる.MRはコルチゾールとアルドステロンの両方に高親和性を示し,脳内のいくつかの部位に発現が認められる.NTSにはMRとともにコルチゾールを不活性なコルチゾンに変換する酵素11β-HSD2が発現しているため,NTSではMRはアルドステロンによって活性化されている85).NTSからはSFOやPVN, RVLMに連絡があり,NTSでのMRの活性化は塩分の摂取を促すとともに血圧上昇を招く86).
2)アンジオテンシンII
AngIIの受容体であるAT1Rは,扁桃体(Am),分界条床核(BNST),視床下部前野(anterior hypothalamic area:AHA),PVN等のストレス反応に関係する脳のさまざまな領域に分布している87–89).これらの部位は脳室周囲器官であるSFOやOVLTに連絡がある(図5参照).ラットにおいてSFO特異的にAT1Rを阻害すると,拘束ストレスに反応したACTHとコルチコステロンの分泌が低下する90).また,AT1Rを欠損したマウスでは嫌悪ストレスに応じた心臓・血管系の反応が低下している91).
免疫系がストレスによる高血圧発症に関与していることは,リンパ球を欠くRAG-1欠損マウスではストレスによる血圧上昇が認められないこと,これがT細胞の移植によって回復することから明らかになった92).ストレスによる高血圧の発生と血管へのT細胞の浸潤は,AngIIによって著しく促進される92).また低レベルのAngIIを長期間静注すると,ストレスによる脳室周囲器官におけるFosの発現を増強するとともに,腎臓の交感神経活性を増大させることがわかっている93).
ヒトの大腸内容物1 g中には1011~1012個の細菌が存在しており,その種類は2900以上におよぶ.高塩食を摂ると腸内細菌叢に変化が生じることがわかった94, 95).また,DSR, SHR, DOCA-saltモデル等の高血圧を示す動物モデルでは腸内細菌叢に変化が起こっている96–99).脂肪分と塩分の多い西洋食を摂るヒトでは,腸内細菌の多様性が減少しバランス失調が起きている100, 101).同様に,高血圧のヒトとそうでないヒトとの間にも,腸内細菌叢に違いがみられる102–104).さらに,高血圧を示す動物やヒトの腸内細菌の移植によって,正常圧の動物に高血圧を発症させることができる105).
腸管には多くの免疫細胞が存在し,Th1やTh17のT細胞サブセットはIFN-γ, TNFαやIL17-A等の炎症性サイトカインを分泌することが知られるが,これらの炎症誘導性T細胞が高血圧のヒトや動物では増加している106, 107).また,最近,マクロファージ,単球や樹状細胞等の自然免疫系が高血圧の発症に関与していることが言われている108).
また,食事における食物繊維の重要性が指摘されている.難溶性デンプンは小腸では消化されず,大腸常在細菌によって分解され,それらのエネルギー源となるだけでなく,酢酸,プロピオン酸,酪酸などの短鎖脂肪酸を生成する.短鎖脂肪酸は,Naや水の吸収,消化管上皮細胞の増殖,粘液分泌,免疫細胞の制御等に影響を与えることが知られている.食物繊維や酢酸,酪酸は腸内細菌叢に影響を与え,DOCA-saltマウスモデルにおいて血圧を低下させる99, 109).酢酸は睡眠時無呼吸に伴う高血圧の発症を抑制する110).またプロピオン酸や酪酸の長期摂取は,制御性T細胞(Treg)の増殖を助け血圧を低下させるとの報告がある111, 112).
末梢の炎症は中枢を介して交感神経活性を上昇させ,神経性高血圧の発症につながる113).炎症は免疫反応の一つであり,炎症の古典的サインとして,血流の増加,血管透過性の増加,炎症性サイトカインの分泌,白血球の誘導等が挙げられる114).これらのプロセスは転写調節因子NF-κBによって制御されており,その下流でTNF-α, IL-1β, IL-6等の炎症誘導性サイトカインの分泌が活性化される115).
血管内にTNF-αやIL-1βを注入すると,腎臓交感神経活性,心拍数,動脈圧を増加させる.そして,これらの反応はSFOの欠損によって弱められることから,SFOが脳内の炎症性サイトカインの検知部位と考えられる116).TNF-αやIL-1βのSFOへの注入による効果は,SFOをアンジオテンシン変換酵素(ACE)の阻害剤やAngII受容体遮断薬で前処理すると減弱する117).さらに,TNF-αやIL-1βをSFOに注入すると,ACE, AngII受容体,PGE2の合成酵素であるシクロオキシゲナーゼ-2等の炎症性シグナル関連分子のmRNAの発現が,SFOだけでなく下流のPVNにおいても増加し,動脈圧の上昇につながる118, 119).以上まとめると,炎症性サイトカインはレニン-アンジオテンシン系を通してSFO内で働き,下流のPVNへの神経路を活性化して,交感神経活動および血圧を上昇させていると考えられる(図5).
AngIIの慢性的投与で高血圧を誘導すると,PVN内に活性化ミクログリアが出現し,ミクログリアの活性化を抑えるミノサイクリン処理すると高血圧は大きく改善する120).AngIIあるいはLPSを使ってin vitroで活性化したミクログリア細胞(N9細胞)をマウス脳室内に注入すると,上昇血圧の持続時間が延伸する121).ミクログリアの活性化をミノサイクリンで抑えておくとこの現象はみられない121).また,高血圧ラット(SHR)と正常血圧ラット(WKY)間の骨髄移植実験から,骨髄由来の末梢炎症細胞の脳内への侵入が高血圧発症に寄与していることが示された(図6)122).この実験でもミノサイクリンが有効な抑制効果を示す.これらの系でも,中枢の交感神経系の活性が免疫系を介して調節されていると思われる.オメガ3等の多価不飽和脂肪酸は神経炎症を抑制することによって血圧を下げる効果があるといわれている123).
引用文献References
1) Weinberger, M.H. (1996) Salt sensitivity of blood pressure in humans. Hypertension, 27, 481–490.
2) Weinberger, M.H. (2006) Pathogenesis of salt sensitivity of blood pressure. Curr. Hypertens. Rep., 8, 166–170.
3) Rapp, J.P. (1982) Dahl salt-susceptible and salt-resistant rats. A review. Hypertension, 4, 753–763.
4) Okamoto, K. & Aoki, K. (1963) Development of a strain of spontaneously hypertensive rats. Jpn. Circ. J., 27, 282–293.
5) Yemane, H., Busauskas, M., Burris, S.K., & Knuepfer, M.M. (2010) Neurohumoral mechanisms in deoxycorticosterone acetate (DOCA)-salt hypertension in rats. Exp. Physiol., 95, 51–55.
6) Ufnal, M., Drapala, A., Sikora, M., & Zera, T. (2011) Early high-sodium solid diet does not affect sodium intake, sodium preference, blood volume and blood pressure in adult Wistar-Kyoto rats. Br. J. Nutr., 106, 292–296.
7) Gomes, P.M., Sá, R.W.M., Aguiar, G.L., Paes, M.H.S., Alzamora, A.C., Lima, W.G., de Oliveira, L.B., Stocker, S.D., Antunes, V.R., & Cardoso, L.M. (2017) Chronic high-sodium diet intake after weaning lead to neurogenic hypertension in adult Wistar rats. Sci. Rep., 7, 5655.
8) Huang, B.S., Van Vliet, B.N., & Leenen, F.H. (2004) Increases in CSF [Na+] precede the increases in blood pressure in Dahl S rats and SHR on a high-salt diet. Am. J. Physiol. Heart Circ. Physiol., 287, H1160–H1166.
9) Gluckman, P.D. & Hanson, M.A. (2004) Living with the past: evolution, development, and patterns of disease. Sience, 305, 1733–1736.
10) Wenzel, U., Turner, J.E., Krebs, C., Kurts, C., Harrison, D.G., & Ehmke, H. (2016) Immune Mechanisms in Arterial Hypertension. J. Am. Soc. Nephrol., 27, 677–686.
11) Simmonds, S.S., Lay, J., & Stocker, S.D. (2014) Dietary salt intake exaggerates sympathetic reflexes and increases blood pressure variability in normotensive rats. Hypertension, 64, 583–589.
12) Xue, B., Zhang, Z., Johnson, R.F., & Johnson, A.K. (2012) Sensitization of slow pressor angiotensin II (ANG II) initiated hypertension: induction of sensitization by prior AngII treatment. Hypertension, 59, 459–466.
13) Xue, B., Zhang, Z., Roncari, C.F., Guo, F., & Johnson, A.K. (2012) Aldosterone acting through the central nervous system sensitizes angiotensin II-Induced hypertension. Hypertension, 60, 1023–1030.
14) Xue, B., Thunhorst, R.L., Yu, Y., Guo, F., Beltz, T.G., Felder, R.B., & Johnson, A.K. (2016) Central Renin-Angiotensin System Activation and Inflammation Induced by High-Fat Diet Sensitize Angiotensin II-Elicited Hypertension. Hypertension, 67, 163–170.
15) Xue, B., Yu, Y., Zhang, Z., Guo, F., Beltz, T.G., Thunhorst, R.L., Felder, R.B., & Johnson, A.K. (2016) Leptin Mediates High-Fat Diet Sensitization of Angiotensin II-Elicited Hypertension by Upregulating the Brain Renin-Angiotensin System and Inflammation. Hypertension, 67, 970–976.
16) Clayton, S.C., Zhang, Z., Beltz, T., Xue, B., & Johnson, A.K. (2014) CNS neuroplasticity and salt-sensitive hypertension induced by prior treatment with subpressor doses of ANG II or aldosterone. Am. J. Physiol. Regul. Integr. Comp. Physiol., 306, R908–R917.
17) Johnson, A.K., Zhang, Z., Clayton, S.C., Beltz, T.G., Hurley, S.W., Thunhorst, R.L., & Xue, B. (2015) The roles of sensitization and neuroplasticity in the long-term regulation of blood pressure and hypertension. Am. J. Physiol. Regul. Integr. Comp. Physiol., 309, R1309–R1325.
18) Watanabe, E., Fujikawa, A., Matsunaga, H., Yasoshima, Y., Sako, N., Yamamoto, T., Saegusa, C., & Noda, M. (2000) Nav2/NaG channel is involved in control of salt-intake behavior in the CNS. J. Neurosci., 20, 7743–7751.
19) Goldin, A.L., Barchi, R.L., Caldwell, J.H., Hofmann, F., Howe, J.R., Hunter, J.C., Kallen, R.G., Mandel, G., Meisler, M.H., Netter, Y.B., et al. (2000) Nomenclature of voltage-gated sodium channels. Neuron, 28, 365–368.
20) Hiyama, T.Y., Watanabe, E., Ono, K., Inenaga, K., Tamkun, M.M., Yoshida, S., & Noda, M. (2002) Nax is involved in the sodium level sensing in the CNS. Nat. Neurosci., 5, 511–512.
21) Watanabe, E., Hiyama, T.Y., Kodama, R., & Noda, M. (2002) Nax sodium channel is expressed in non-myelinating Schwann cells and alveolar type II cells in mice. Neurosci. Lett., 330, 109–113.
22) Hiyama, T.Y., Watanabe, E., Okado, H., & Noda, M. (2004) The subfornical organ is the primary locus of sodium-level sensing by Nax sodium channels for the control of salt-intake behavior. J. Neurosci., 24, 9276–9281.
23) Noda, M. & Hiyama, T.Y. (2005) Sodium-level-sensitive sodium channel and salt-intake behavior. Chem. Senses, 30(Supple. 1), i44–i45.
24) Noda, M. (2006) The subfornical organ, a specialized sodium channel, and the sensing of sodium levels in the brain. Neuroscientist, 12, 80–91.
25) Watanabe, E., Hiyama, T.Y., Shimizu, H., Kodama, R., Hayashi, N., Miyata, S., Yanagawa, Y., Obata, K., & Noda, M. (2005) Sodium-level-sensitive sodium channel Nax is expressed in glial laminate processes in the sensory circumventricular organs. Am. J. Physiol. Regul. Integr. Comp. Physiol., 290, R568–R576.
26) Shimizu, H., Watanabe, E., Hiyama, T.Y., Nagakura, A., Fujikawa, A., Okado, H., Yanagawa, Y., Obata, K., & Noda, M. (2007) Glial Nax channels control lactate signaling to neurons for brain [Na+] sensing. Neuron, 54, 59–72.
27) Noda, M. (2007) Hydromineral neuroendocrinology: mechanism of sensing sodium levels in the mammalian brain. Exp. Physiol., 92, 513–522.
28) Nagakura, A., Hiyama, T.Y., & Noda, M. (2010) Nax-deficient mice show normal vasopression response to dehydration. Neurosci. Lett., 472, 161–165.
29) Hiyama, T.Y., Matsuda, S., Fujikawa, A., Matsumoto, M., Watanabe, E., Kajiwara, H., Niimura, F., & Noda, M. (2010) Autoimmunity to the sodium-level sensor in the brain causes essential hypernatremia. Neuron, 66, 508–522.
30) Matsumoto, M., Fujikawa, A., Suzuki, R., Shimizu, H., Kuboyama, K., Hiyama, T.Y., Hall, R.A., & Noda, M. (2012) SAP97 promotes the stability of Nax channels at the plasma membrane. FEBS Lett., 586, 3805–3812.
31) Hiyama, T.Y., Yoshida, M., Matsumoto, M., Suzuki, R., Matsuda, T., Watanabe, E., & Noda, M. (2013) Endothelin-3 Expression in the Subfornical Organ Enhancesx the Sensitivity of Nax, the Brain Sodium-Level Sensor, to Suppress Salt Intake. Cell Metab., 17, 507–519.
32) Noda, M. & Sakuta, H. (2013) Central regulation of body-fluid homeostasis. Trends Neurosci., 36, 661–673.
33) Noda, M. & Hiyama, T.Y. (2015) The Nax Channel: What It Is and What It Does. Neuroscientist, 21, 399–412.
34) Noda, M. & Hiyama, T.Y. (2015) Sodium sensing in the brain. Pflugers Arch., 467, 465–474.
35) Sakuta, H., Nishihara, E., Hiyama, T.Y., Lin, C.H., & Noda, M. (2016) Nax signaling evoked by an increase in [Na+] inCSF induces water intake via EET-mediated TRPV4 activation. Am. J. Physiol. Regul. Integr. Comp. Physiol., 311, R299–R306.
36) Hiyama, T.Y. & Noda, M. (2016) Sodium sensing in the subfornical organ and body-fluid homeostasis. Neurosci. Res., 113, 1–11.
37) Hiyama, T.Y., Utsunomiya, A.N., Matsumoto, M., Fujikawa, A., Lin, C.H., Hara, K., Kagawa, R., Okada, S., Kobayashi, M., Anzo, M., et al. (2016) Adipsic hypernatremia without hypothalamic lesions accompanied by autoantibodies to subfornical organ. Brain Pathol., 27, 323–331.
38) Matsuda, T., Hiyama, T.Y., Niimura, F., Matsusaka, T., Fukamizu, A., Kobayashi, K., Kobayashi, K., & Noda, M. (2017) Distinct neural mechanisms for the control of thirst and salt appetite in the subfornical organ. Nat. Neurosci., 20, 230–241.
39) Utsunomiya, N.A., Hiyama, T.Y., Okada, S., Noda, M., & Kobayashi, M. (2017) Characteristic clinical features of adipsic hypernatremia patients with subfornical organ-targeting antibody. Clin. Pediatr. Endocrinol., 26, 197–205.
40) Sakuta, H., Lin, C.-H., Yamada, M., Kita, Y., Tokuoka, S.M., Shimizu, T., & Noda, M. (2019) Nax-positive glial cells in the organum vasculosum laminae terminalis produce epoxyeicosatrienoic acids to induce water intake in response to increases in [Na+] in body fluids. Neurosci. Res., in press.
41) Nomura, K., Hiyama, T.Y., Sakuta, H., Matsuda, T., Lin, C.-H., Kobayashi, K., Kobayashi, K., Kuwaki, T., Takahashi, K., Matsui, S., et al. (2018) [Na+] increases in body fluids sensed by central Nax induce sympathetically mediated blood pressure elevations via H+-dependent activation of ASIC1a. Neuron, 101, 60–75.
42) Lim, K., Jackson, K.L., Sata, Y., & Head, G.A. (2017) Factors Responsible for Obesity-Related Hypertension. Curr. Hypertens. Rep., 19, 53.
43) Muntzel, M.S., Al-Naimi, O.A., Barclay, A., & Ajasin, D. (2012) Cafeteria diet increases fat mass and chronically elevates lumbar sympathetic nerve activity in rats. Hypertension, 60, 1498–1502.
44) Armitage, J.A., Burke, S.L., Prior, L.J., Barzel, B., Eikelis, N., Lim, K., & Head, G.A. (2012) Rapid onset of renal sympathetic nerve activation in rabbits fed a high-fat diet. Hypertension, 60, 163–171.
45) Lim, K., Burke, S.L., & Head, G.A. (2013) Obesity-related hypertension and the role of insulin and leptin in high-fat-fed rabbits. Hypertension, 61, 628–634.
46) Luckett, B.S., Frielle, J.L., Wolfgang, L., & Stocker, S.D. (2013) Arcuate nucleus injection of an anti-insulin affibody prevents the sympathetic response to insulin. Am. J. Physiol. Heart Circ. Physiol., 304, H1538–H1546.
47) Shi, Z., Li, B., & Brooks, V.L. (2015) Role of the Paraventricular Nucleus of the Hypothalamus in the Sympathoexcitatory Effects of Leptin. Hypertension, 66, 1034–1041.
48) Harlan, S.M., Morgan, D.A., Agassandian, K., Guo, D.F., Cassell, M.D., Sigmund, C.D., Mark, A.L., & Rahmouni, K. (2011) Ablation of the leptin receptor in the hypothalamic arcuate nucleus abrogates leptin-induced sympathetic activation. Circ. Res., 108, 808–812.
49) Rahmouni, K., Haynes, W.G., Morgan, D.A., & Mark, A.L. (2003) Role of melanocortin-4 receptors in mediating renal sympathoactivation to leptin and insulin. J. Neurosci., 23, 5998–6004.
50) Ward, K.R., Bardgett, J.F., Wolfgang, L., & Stocker, S.D. (2011) Sympathetic response to insulin is mediated by melanocortin 3/4 receptors in the hypothalamic paraventricular nucleus. Hypertension, 57, 435–441.
51) Shi, Z., Madden, C.J., & Brooks, V.L. (2017) Arcuate neuropeptide Y inhibits sympathetic nerve activity via multiple neuropathways. J. Clin. Invest., 127, 2868–2880.
52) Cassaglia, P.A., Shi, Z., Li, B., Reis, W.L., Clute-Reinig, N.M., Stern, J.E., & Brooks, V.L. (2014) Neuropeptide Y acts in the paraventricular nucleus to suppress sympathetic nerve activity and its baroreflex regulation. J. Physiol., 592, 1655–1675.
53) Cassaglia, P.A., Shi, Z., & Brooks, V.L. (2016) Insulin increases sympathetic nerve activity in part by suppression of tonic inhibitory neuropeptide Y inputs into the paraventricular nucleus in female rats. Am. J. Physiol. Regul. Integr. Comp. Physiol., 311, R97–R103.
54) Lim, K., Barzel, B., Burke, S.L., Armitage, J.A., & Head, G.A. (2016) Origin of Aberrant Blood Pressure and Sympathetic Regulation in Diet-Induced Obesity. Hypertension, 68, 491–500.
55) Simonds, S.E., Pryor, J., Ravussin, E., Greenway, F.L., Dileone, R., Allen, A.M., Bassi, J., Elmquist, J.K., Keogh, J.M., Henning, E., et al. (2014) Leptin Mediates the Increase in Blood Pressure Associated with Obesity. Cell, 159, 1404–1416.
56) Young, C.N., Morgan, D.A., Butler, S.D., Mark, A.L., & Davisson, R.L. (2013) The brain subfornical organ mediates leptin-induced increases in renal sympathetic activity but not its metabolic effects. Hypertension, 61, 737–744.
57) Brown, R.J., Meehan, C.A., & Gorden, P. (2015) Leptin Does Not Mediate Hypertension Associated With Human Obesity. Cell, 162, 465–466.
58) Reilly, S.M. & Saltiel, A.R. (2017) Adapting to obesity with adipose tissue inflammation. Nat. Rev. Endocrinol., 13, 633–643.
59) Frühbeck, G. (2004) The adipose tissue as a source of vasoactive factors. Curr. Med. Chem. Cardiovasc. Hematol. Agents, 2, 197–208.
60) Liu, R. & Nikolajczyk, B.S. (2019) Tissue Immune Cells Fuel Obesity-Associated Inflammation in Adipose Tissue and Beyond. Front. Immunol., 10, 1587.
61) Skaper, S.D., Facci, L., Zusso, M., & Giusti, P. (2018) An Inflammation-Centric View of Neurological Disease: Beyond the Neuron. Front. Cell. Neurosci., 12, 72.
62) Sunderram, J. & Androulakis, I.P. (2012) Molecular mechanisms of chronic intermittent hypoxia and hypertension. Crit. Rev. Biomed. Eng., 40, 265–278.
63) McBryde, F.D., Abdala, A.P., Hendy, E.B., Pijacka, W., Marvar, P., Moraes, D.J., Sobotka, P.A., & Paton, J.F. (2013) The carotid body as a putative therapeutic target for the treatment of neurogenic hypertension. Nat. Commun., 4, 2395.
64) Del Rio, R., Moya, E.A., & Iturriaga, R. (2010) Carotid body and cardiorespiratory alterations in intermittent hypoxia: the oxidative link. Eur. Respir. J., 36, 143–150.
65) Peng, Y.J., Yuan, G., Khan, S., Nanduri, J., Makarenko, V.V., Reddy, V.D., Vasavda, C., Kumar, G.K., Semenza, G.L., & Prabhakar, N.R. (2014) Regulation of hypoxia-inducible factor-α isoforms and redox state by carotid body neural activity in rats. J. Physiol., 592, 3841–3858.
66) Irwin, M.R., Carrillo, C., & Olmstead, R. (2014) Sleep loss activates cellular markers of inflammation: Sex differences. Brain Behav. Immun., 24, 54–57.
67) Meier-Ewert, H.K., Ridker, P.M., Rifai, N., Regan, M.M., Price, N.J., Dinges, D.F., & Mullington, J.M. (2004) Effect of sleep loss on C-Reactive protein, an inflammatory marker of cardiovascular risk. J. Am. Coll. Cardiol., 43, 678–683.
68) Waki, H., Liu, B., Miyake, M., Katahira, K., Murphy, D., Kasparov, S., & Paton, J.F.R. (2007) Junctional adhesion molecule-1 is upregulated in spontaneously hypertensive Rats: evidence for a prohypertensive role within the brain stem. Hypertension, 49, 1321–1327.
69) Berger, A.J. (1980) The distribution of the Cat’s carotid sinus nerve afferent and efferent cell bodies using the horseradish peroxidase technique. Brain Res., 190, 309–320.
70) Finley, J.C.W. & Katz, D.M. (1992) The central organization of carotid body afferent projections to the brainstem of the rat. Brain Res., 572, 108–116.
71) Knight, W.D., Little, J.T., Carreno, F.R., Toney, G.M., Mifflin, S.W., & Cunningham, J.T. (2011) Chronic intermittent hypoxia increases blood pressure and expression of FosB/DeltaFosB in central autonomic regions. Am. J. Physiol. Regul. Integr. Comp. Physiol., 301, R131–R139.
72) Bathina, C.S., Rajulapati, A., Franzke, M., Yamamoto, K., Cunningham, J.T., & Mifflin, S. (2013) Knockdown of tyrosine hydroxylase in the nucleus of the solitary tract reduces elevated blood pressure during chronic intermittent hypoxia. Am. J. Physiol. Regul. Integr. Comp. Physiol., 305, R1031–R1039.
73) Sharpe, A.L., Calderon, A.S., Andrade, M.A., Cunningham, J.T., Mifflin, S.W., & Toney, G.M. (2013) Chronic intermittent hypoxia increases sympathetic control of blood pressure: role of neuronal activity in the hypothalamic paraventricular nucleus. Am. J. Physiol. Regul. Integr. Comp. Physiol., 305, H1772–H1780.
74) An, Q. & En-Sheng, Ji. (2014) The nervous mechanism of sympathetic over activity induced by chronic intermittent hypoxia. J. Pulm. Respir. Med., 8, 451.
75) Daulatzai, M.A. (2012) Pathogenesis of Cognitive Dysfunction in Patients with Obstructive Sleep Apnea: A Hypothesis with Emphasis on the Nucleus Tractus Solitarius. Sleep Disord., 2012, 1–18.
76) Esler, M., Eikelis, N., Schlaich, M., Lambert, G., Alvarenga, M., Dawood, T., Kaye, D., Barton, D., Pier, C., Guo, L., et al. (2008) Chronic mental stress is a cause of essential hypertension: presence of biological markers of stress. Clin. Exp. Pharmacol. Physiol., 35, 498–502.
77) Davern, P.J. & Head, G.A. (2011) Role of the medial amygdala in mediating responses to aversive stimuli leading to hypertension. Clin. Exp. Pharmacol. Physiol., 38, 136–143.
78) Vinson, G.P. (2007) Angiotensin II, corticosteroids, type II diabetes and the metabolic syndrome. Med. Hypotheses, 68, 1200–1207.
79) Matthews, K., Schwartz, J., Cohen, S., & Seeman, T. (2006) Diurnal cortisol decline is related to coronary calcification. Psychosom. Med., 68, 657–661.
80) Garg, R. & Adler, G.K.(2009) in Textbook of Nephro-Endocrinology (Singh, A. & Williams G. eds), Elsevier, Burlington, MA.
81) Clamage, D.M., Sanford, C.S., Vander, A.J., & Mouw, D.R. (1976) Effects of psychosocial stimuli on plasma renin activity in rats. Am. J. Physiol., 231, 1290–1294.
82) Carrasco, G.A. & Van de Kar, L.D. (2003) Neuroendocrine pharmacology of stress. Eur. J. Pharmacol., 463, 235–272.
83) Johnson, A.K. & Grippo, A.J. (2006) Sadness and broken hearts: neurohumoral mechanisms and co-morbidity of ischemic heart disease and psychological depression. J. Physiol. Pharmacol., 57(Suppl 11), 5–29.
84) Hlavacova, N. & Jezova, D. (2008) Chronic treatment with the mineralocorticoid hormone aldosterone results in increased anxiety-like behavior. Horm. Behav., 54, 90–97.
85) Funder, J.W. (2009) Reconsidering the roles of the mineralocorticoid receptor. Hypertension, 53, 286–290.
86) Geerling, J.C., Engeland, W.C., Kawata, M., & Loewy, A.D. (2006) Aldosterone target neurons in the nucleus tractus solitarius drive sodium appetite. J. Neurosci., 26, 411–417.
87) Aguilera, G., Kiss, A., Luo, X., & Akbasak, B.S. (1995) The renin angiotensin system and the stress response. Ann. N. Y. Acad. Sci., 771(1 Stress), 173–186.
88) von Bohlen und Halbach, O. & Albrecht, D. (1998) Mapping of angiotensin AT1 receptors in the rat limbic system. Regul. Pept., 78, 51–56.
89) Yang, R.-H., Jin, H., Chen, S.J., Wyss, J.M., & Oparil, S. (1992) Blocking hypothalamic AT1 receptors lowers blood pressure in salt-sensitive rats. Hypertension, 20, 755–762.
90) Krause, E.G., de Kloet, A.D., Scott, K.A., Flak, J.N., Jones, K., Smeltzer, M.D., Ulrich-Lai, Y.M., Woods, S.C., Wilson, S.P., Reagan, L.P., et al. (2011) Blood-Borne Angiotensin II Acts in the Brain to Influence Behavioral and Endocrine Responses to Psychogenic Stress. J. Neurosci., 31, 15009–15015.
91) Davern, P.J., Chen, D., Head, G.A., Chavez, C.A., Walther, T., & Mayorov, D.N. (2009) Role of Angiotensin II Type 1A Receptors in Cardiovascular Reactivity and Neuronal Activation After Aversive Stress in Mice. Hypertension, 54, 1262–1268.
92) Marvar, P.J., Vinh, A., Thabet, S., Lob, H.E., Geem, D., Ressler, K.J., & Harrison, D.G. (2012) T lymphocytes and vascular inflammation contribute to stress-dependent hypertension. Biol. Psychiatry, 71, 774–782.
93) Moretti, J.L., Burke, S.L., Davern, P.J., Evans, R.G., Lambert, G.W., & Head, G.A. (2012) Renal sympathetic activation from long-term low-dose angiotensin II infusion in rabbits. J Hyotertens., 30, 551–560.
94) Wilck, N., Matus, M.G., Kearney, S.M., Olesen, S.W., Forslund, K., Bartolomaeus, H., Haase, S., Mähler, A., Balogh, A., Markó, L., et al. (2017) Salt-responsive gut commensal modulates TH17 axis and disease. Nature, 551, 585–589.
95) Ferguson, J.F., Aden, L.A., Barbaro, N.R., Van Beusecum, J.P., Xiao, L., Simmons, A.J., Warden, C., Pasic, L., Himmel, L.E., Washington, M.K., et al. (2019) High dietary salt–induced DC activation underlies microbial dysbiosis-associated hypertension. JCI Insight, 4, e126241.
96) Yang, T., Santisteban, M.M., Rodriguez, V., Li, E., Ahmari, N., Carvajal, J.M., Zadeh, M., Gong, M., Qi, Y., Zubcevic, J., et al. (2015) Gut dysbiosis is linked to hypertension. Hypertension, 65, 1331–1340.
97) Adnan, S., Nelson, J.W., Ajami, N.J., Venna, V.R., Petrosino, J.F., Bryan, R.M. Jr., & Durgan, D.J. (2017) Alterations in the gut microbiota can elicit hypertension in rats. Physiol. Genomics, 49, 96–104.
98) Mell, B., Jala, V.R., Mathew, A.V., Byun, J., Waghulde, H., Zhang, Y., Haribabu, B., Vijay-Kumar, M., Pennathur, S., & Joe, B. (2015) Evidence for a link between gut microbiota and hypertension in the Dahl rat. Physiol. Genomics, 47, 187–197.
99) Marques, F.Z., Nelson, E., Chu, P.Y., Horlock, D., Fiedler, A., Ziemann, M., Tan, J.K., Kuruppu, S., Rajapakse, N.W., El-Osta, A., et al. (2017) High-Fiber Diet and Acetate Supplementation Change the Gut Microbiota and Prevent the Development of Hypertension and Heart Failure in Hypertensive Mice. Circulation, 135, 964–977.
100) Senghor, B., Sokhna, C., Ruimy, R., & Lagier, J.C. (2018) Gut microbiota diversity according to dietary habits and geographical provenance. Human Microbiome Journal, 7-8, 1–9.
101) Jama, H.A., Beale, A., Shihata, W.A., & Marques, F.Z. (2019) The effect of diet on hypertensive pathology: is there a link via gut microbiota-driven immunometabolism? Cardiovasc. Res., 115, 1435–1447.
102) Li, J., Zhao, F., Wang, Y., Chen, J., Tao, J., Tian, G., Wu, S., Liu, W., Cui, Q., Geng, B., et al. (2017) Gut microbiota dysbiosis contributes to the development of hypertension. Microbiome, 5, 14.
103) Yan, Q., Gu, Y., Li, X., Yang, W., Jia, L., Chen, C., Han, X., Huang, Y., Zhao, L., Li, P., et al. (2017) Alterations of the Gut Microbiome in Hypertension. Front. Cell. Infect. Microbiol., 7, 381.
104) Dan, X., Mushi, Z., Baili, W., Han, L., Enqi, W., Huanhu, Z., & Shuchun, L. (2019) Differential Analysis of Hypertension-Associated Intestinal Microbiota. Int. J. Med. Sci., 16, 872–881.
105) Li, J., Zhao, F., Wang, Y., Chen, J., Tao, J., Tian, G., Wu, S., Liu, W., Cui, Q., Geng, B., et al. (2017) Gut microbiota dysbiosis contributes to the development of hypertension. Microbiome, 5, 14.
106) Luo, T., Ji, W.J., Yuan, F., Guo, Z.Z., Li, Y.X., Dong, Y., Ma, Y.Q., Zhou, X., & Li, Y.M. (2016) Th17/Treg Imbalance Induced by Dietary Salt Variation Indicates Inflammation of Target Organs in Humans. Sci. Rep., 6, 26767.
107) Wilck, N., Matus, M.G., Kearney, S.M., Olesen, S.W., Forslund, K., Bartolomaeus, H., Haase, S., Mähler, A., Balogh, A., Markó, L., et al. (2017) Salt-responsive gut commensal modulates TH17 axis and disease. Nature, 551, 585–589.
108) Jama, H.A., Beale, A., Shihata, W.A., & Marques, F.Z. (2019) The effect of diet on hypertensive pathology: is there a link via gut microbiota-driven immunometabolism? Cardiovasc. Res., 115, 1435–1447.
109) Wang, L., Zhu, Q., Lu, A., Liu, X., Zhang, L., Xu, C., Liu, X., Li, H., & Yang, T. (2017) Sodium butyrate suppresses angiotensin II-induced hypertension by inhibition of renal (pro)renin receptor and intrarenal renin-angiotensin system. J. Hypertens., 35, 1899–1908.
110) Ganesh, B.P., Nelson, J.W., Eskew, J.R., Ganesan, A., Ajami, N.J., Petrosino, J.F., Bryan, R.M. Jr., & Durgan, D.J. (2018) Prebiotics, Probiotics, and Acetate Supplementation Prevent Hypertension in a Model of Obstructive Sleep Apnea. Hypertension, 72, 1141–1150.
111) Bartolomaeus, H., Balogh, A., Yakoub, M., Homann, S., Markó, L., Höges, S., Tsvetkov, D., Krannich, A., Wundersitz, S., Avery, E.G., et al. (2019) Short-Chain Fatty Acid Propionate Protects From Hypertensive Cardiovascular Damage. Circulation, 139, 1407–1421.
112) Kim, S., Goel, R., Kumar, A., Qi, Y., Lobaton, G., Hosaka, K., Mohammed, M., Handberg, E.M., Richards, E.M., Pepine, C.J., et al. (2018) Imbalance of gut microbiome and intestinal epithelial barrier dysfunction in patients with high blood pressure. Clin. Sci. (Lond.), 132, 701–718.
113) Mowry, F.E. & Biancardi, V.C. (2019) Neuroinflammation in hypertension: the renin-angiotensin system versus pro-resolution pathways. Pharmacol. Res., 144, 279–291.
114) Tian, R., Hou, G., Li, D., & Yuan, T.F. (2014) A possible change process of inflammatory cytokines in the prolonged chronic stress and its ultimate implications for health. ScientificWorldJournal, 2014, 780616.
115) Shih, R.H., Wang, C.Y., & Yang, C.M. (2015) NF-kappaB Signaling Pathways in Neurological Inflammation: A Mini Review. Front. Mol. Neurosci., 8, 77.
116) Wei, S.G., Zhang, Z.H., Beltz, T.G., Yu, Y., Johnson, A.K., & Felder, R.B. (2013) Subfornical organ mediates sympathetic and hemodynamic responses to blood-borne proinflammatory cytokines. Hypertension, 62, 118–125.
117) Wei, S.G., Yu, Y., Zhang, Z.H., & Felder, R.B. (2015) Proinflammatory cytokines upregulate sympathoexcitatory mechanisms in the subfornical organ of the rat. Hypertension, 65, 1126–1133.
118) Bardgett, M.E., Holbein, W.W., Herrera-Rosales, M., & Toney, G.M. (2014) Ang II-salt hypertension depends on neuronal activity in the hypothalamic paraventricular nucleus but not on local actions of tumor necrosis factor-α. Hypertension, 63, 527–534.
119) Sriramula, S., Cardinale, J.P., & Francis, J. (2013) Inhibition of TNF in the brain reverses alterations in RAS components and attenuates angiotensin II-induced hypertension. PLoS One, 8, e63847.
120) Shi, P., Diez-Freire, C., Jun, J.Y., Qi, Y., Katovich, M.J., Li, Q., Sriramula, S., Francis, J., Sumners, C., & Raizada, M.K. (2010) Brain microglial cytokines in neurogenic hypertension. Hypertension, 56, 297–303.
121) Shen, X.Z., Li, Y., Li, L., Shah, K.H., Bernstein, K.E., Lyden, P., & Shi, P. (2015) Microglia participate in neurogenic regulation of hypertension. Hypertension, 66, 309–316.
122) Santisteban, M.M., Ahmari, N., Carvajal, J.M., Zingler, M.B., Qi, Y., Kim, S., Joseph, J., Garcia-Pereira, F., Johnson, R.D., Shenoy, V., et al. (2015) Involvement of bone marrow cells and neuroinflammation in hypertension. Circ. Res., 117, 178–191.
123) Mowry, F.E. & Biancardi, V.C. (2019) Neuroinflammation in hypertension: the renin-angiotensin system versus pro-resolution pathways. Pharmacol. Res., 144, 279–291.