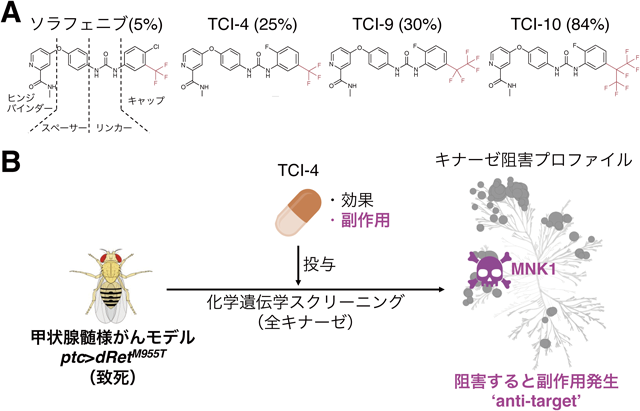
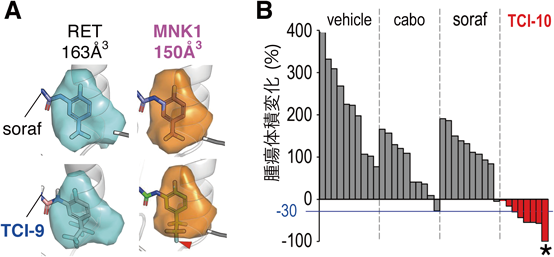
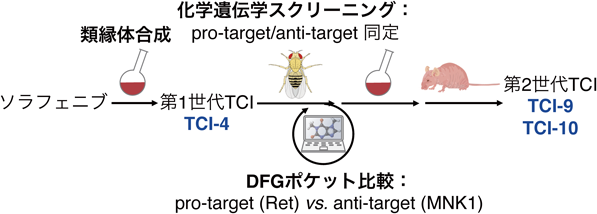
個体を用いた新規抗がん剤創薬基盤~既存薬の合理的改変手法の確立~A whole-animal platform for developing novel anti-cancer leads
北海道大学遺伝子病制御研究所がん制御学分野Division of Biomedical Oncology, Hokkaido University Institute for Genetic Medicine ◇ 〒060–0815 札幌市北区北15条西7丁目 ◇ Kita-15 Nishi-7, Kita-ku, Sapporo, Hokkaido 060–0815, Japan
発行日:2020年8月25日Published: August 25, 2020