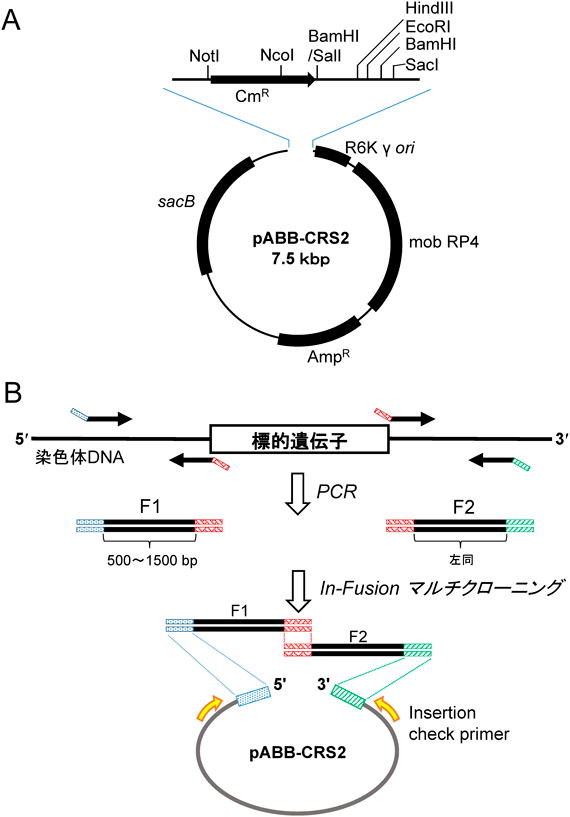
今回紹介するスーサイドベクターpABB-CRS2を用いて遺伝子を欠損させる方法は,阿部章夫教授(北里大)の欠損株作製法をもとに改良したものである5).基本的には,大腸菌SM10 λpir株6)と接合が可能なグラム陰性菌であるなら,菌種を問わず遺伝子欠損は可能である(Bordetella属,Escherichia属,Proteus属,Salmonella属,Vibrio属,Yersinia属などで報告がある).
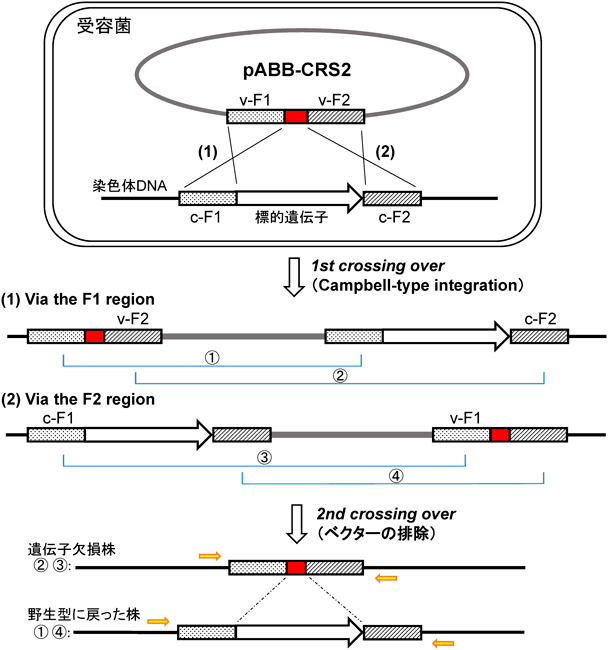
以下,遺伝子を任意の約20塩基対に置換することで,目的の遺伝子の機能を欠損させるという最もシンプルな欠損方法を示す.まず欠損させたい遺伝子の前後の配列を,染色体DNAを鋳型にしてPCRで増幅する.その二つのDNA断片を,In-Fusion法を用いてpABB-CRS2にマルチクローニングする(図1).このベクターは,R6Kプラスミド由来DNA複製起点を持っているので,pir遺伝子を持つ大腸菌SM10 λpir株のような菌株でしか複製することができない(複製開始には,その起点領域にpir遺伝子から発現するπタンパク質の結合が必須なため)7).さらに,接合伝達に必要なmob RP4領域,スクロース選択時に必要なsacB遺伝子そしてアンピシリン(Amp)耐性遺伝子を含んでいる(図1A).クローニングされたpABB-CRS2をSM10 λpir株に導入し,遺伝子を欠損させたい受容菌と混ぜて接合(conjugation)させる6).このベクターはmob RP4領域を持つので,SM10 λpir株由来の接合伝達システム(RP4)を介して受容菌に移動し,受容菌の染色体DNAと相同な配列(図2におけるF1あるいはF2)で一度目の相同組換え(Site-specific recombination, Campbell-type integration)を起こす(1st crossing over)8).この段階では,染色体への組み込まれ方は二つのパターンが存在する(図2).
実験では,このベクターにおける「pir遺伝子産物がないと染色体外では複製できない」そして「Amp耐性遺伝子を持つ」という特徴を利用して,Amp存在下のM9培地で培養することでベクターが染色体に組み込まれた受容菌株のみを選択する.次に,この菌株の染色体からベクター全体を除くため,5%スクロースを含むLB培地で培養する.ベクターにコードされているSacBタンパク質は,スクロース存在下でグラム陰性菌の生育に有害な物質を産生することで知られる[レバン(levan,多糖)が大量に合成されペリプラズムに蓄積し,溶菌に至る]9).よって,SacBが存在している菌はスクロース存在下では生育できないので,二度目の相同組換えを起こし(2nd crossing over),ベクターを排除した受容菌株のみが生育可能となる(図2).このように,ベクターが細胞内に存在するとその細菌は生育できないので,スーサイド(suicide,自滅)ベクターと呼ばれる.
ベクターの排除によって,結果として,目的の遺伝子が欠損した菌株と元の野生株に戻った菌株を得ることになる(図2).この二つを区別するために,欠損する対象の領域を挟んでコロニーPCRをして,その産物の長さを比較することで欠損株を同定する.
示した日数は,大腸菌の遺伝子欠損に関して,最短で行えた場合の目安である.菌種や欠損する遺伝子によって増殖の速度が異なるので,コロニーの成長を注意して観察し培養時間を調整すべきである.表1に,株の種類,ベクター含有の有無および培地の種類との増殖能の関係をまとめた.この表を参考にしながら読むと,実験操作の「意味」の迅速な理解につながるであろう.また,「脚注」には,実際に実験する上でのコツも書かれているので,この点においても参考にしていただきたい.
表1 供与菌(SM10 λpir)および受容菌の培地による増殖能の比較| 培地の種類 | SM10 λpir | 受容菌 |
|---|
| vを含まない | vを含む | vを含まない(接合なし)(野生株) | ゲノムにvが組み込まれた状態 | vが排出された状態(欠損株) |
|---|
| LB (−Amp) | ○ | ○ | ○ | ○ | ○ |
| LB (+Amp) | × | ○ | × | ○ | × |
| M9 (+Amp) | × | × | × | ○ | × |
| LB (+Sc, −NaCl) | ○ | × | ○ | × | ○ |
| ○:増殖可,×:増殖不可,v:スーサイドベクター,Sc:スクロース. |
【1日目:遺伝子欠損用DNA断片の作製,クローニングおよびSM10 λpir株への導入】
1)スーサイドベクターに対し制限酵素処理およびアガロース電気泳動を行い,切断された線状化ベクターを精製しておく(今回はNotIおよびNcoIを使用した).
2)PCRにより欠損させたい遺伝子の上流と下流に対応する二つのDNA断片F1およびF2をそれぞれ作製する.F1およびF2の相同領域(図1B,黒)は,受容菌の相同組換えの効率により,500塩基対から1500塩基対程度とる必要がある(F1とF2は同じ長さにする;大腸菌の場合,1000塩基対程度).使用するプライマーには,ベクターの制限酵素処理後に生じた末端配列に相同な15塩基(図1B,青/ドット,緑/斜線;表2,配列)および,二つの断片をつなぎ合わせるための互いに相同な任意の配列の約20塩基(図1B,赤/モザイク)が付加されている.
表2 本実験で使用したユニバーサルプライマー| In-Fusionのリンカー部分 | 配列 |
|---|
| 5′末端NotI用配列(青) | 5′-GTCACTATGGCGGCC-3′ |
| 3′末端NcoI用配列(緑) | 5′-TAATATTTGCCCATG-3′ |
| Insertion check primer | 配列 |
|---|
| Forward | 5′-GATTTGCAGCATATCATGG-3′ |
| Reverse | 5′-CAGTTCAACCTGTTGATAGTACG-3′ |
3)DNA断片F1およびF2と精製済みの線状化ベクターを混ぜ,In-Fusion法によりマルチクローニングする.その反応液を供与菌であるSM10 λpir株に加え,通常のHeat shock法により形質転換し,LBプレート(+Amp)に播種する.表3に,各実験で使用される培地の組成および作製法をまとめた。
【2日目】
1)プレートに生えたコロニーに対し,Insertion check primer(図1B,黄;表2,配列)を使ってコロニーPCRすることで二つの断片がベクターにクローニングされているかを確認する.
表3 培地の組成および作製方法| ・LB培地 | [通常] | [+5%スクロース,−NaCl] |
| Bactro tryptone | 10 g | 10 g |
| Bactro yeast extract | 5 g | 5 g |
| NaCl | 10 g | — |
| 5N NaOH | 0.2 mL | 0.2 mL |
| スクロース | — | 5 g |
| 純水(RO水) | 1 L | 1 L →オートクレーブ滅菌 |
| アンピシリン入り(+Amp)にする場合は,50°C付近まで冷えてから50 mg/mL(1000×)のAmpを1 mL加える. |
| ・10×M9 salt |
| Na2HPO4 | 60 g |
| KH2PO4 | 30 g |
| NH4Cl | 10 g |
| NaCl | 5 g →純水(RO水)で1 Lにメスアップし,オートクレーブ滅菌 |
| ・M9プレート(+Amp):10枚分 |
| (a) 10×M9 salt | 20 mL |
| 1 M MgSO4 | 200 µL |
| (b)グルコース | 0.4 g |
| Agar | 3 g |
| 純水(RO水) | 180 mL |
| (a)および(b)の溶液を別々にオートクレーブ滅菌し,50°C付近まで冷えてから混合する.最後に50 mg/mL(1000×)のAmpを200 µL加え,プレートに分注する. |
2)遺伝子欠損用のベクターの導入が確認された大腸菌SM10 λpir株を5 mLのLB液体培地(+Amp)および,遺伝子を欠損させたい菌(今回は大腸菌常在株を使用,以下,受容菌と呼ぶ)を5 mLのLB液体培地(−Amp)で,それぞれ一晩,振とう培養(130~160 rpm)する.以下,保温温度はすべて37°Cである.
【3日目:1st crossing over】
1)SM10 λpir株と受容菌の培養液から200 µLを採取し,それぞれ別々の5 mLのLB液体培地(−Amp)に加え,3 h振とう培養する.
2)それぞれの培養液から200 µLを採取し,マイクロチューブに入れて混和し,3 h静置することで,受容菌の染色体DNAに欠損用ベクターを接合伝達させる.
3)マイクロチューブを室温で5000 rpm, 10 min遠心し,上清をデカンテーションで取り除く.
4)ペレット状となった菌体を1 mLの滅菌済み生理食塩水でよく懸濁し,再度3)を繰り返す.その後,ペレット状の菌体を200~500 µLの滅菌済み生理食塩水で懸濁し,そのうち100 µLをM9プレート(+Amp)に播種し,一晩培養する.M9培地では,アミノ酸要求株であるSM10 λpir株は増殖できないので,受容菌の染色体DNA内に欠損用ベクターが組み込まれた菌株のみがAmpで選択される.
【4日目】
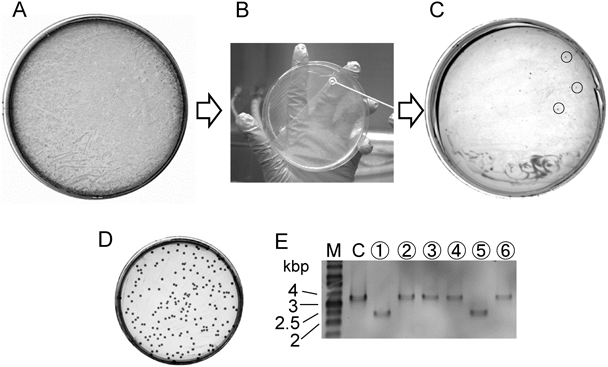
プレートに厚く広がって生えてきた菌体(図3A)をディスポループ(あるいは,白金耳)ですべてかきとり(図3B),新たなM9プレート(+Amp)の全体になるべく薄く塗りつけ,再度,一晩培養する.
Key point: 3日目のプレートのコロニー(らしきもの)を使って実験を進めても,本来は増殖できない受容菌だったりSM10 λpirだったりする可能性がとても高い(あるいはそれらの混合物).これは,LB培地の持ち込みだったり,播種のとき菌体の密度が高いため菌体どうしの排出物を使ったりして,本来増殖できない菌体が増殖してしまったと考えられる(つまり,negativeなコロニーが全体に生えてバックグラウンドが高くなり,本来のpositiveなコロニーが採取できにくくなっている).そのため,再度,播種し直すことで,positiveなコロニーの選択性を高める.
【5日目(夜):2nd crossing over】
コロニーの成長が遅いので,できるだけ培養時間をとる.しかし,十分時間をかけても一定以上大きくならないので(小さいままなので),むだに時間をかけないように注意する.採取可能なコロニーとなったら(図3C),2~3個コロニーをとって,それぞれ特別なLB液体培地(+5%スクロース,−NaCl)で10~16 h培養する.
この過程で,培地にスクロースが存在するため,染色体DNAからベクターを排除した(二度目の相同組換えを起こした)受容菌株のみ選択される.
【6日目】
培養液をLB液体培地(+5%スクロース,−NaCl)で,シングルコロニーになるように薄めた後(~10−4)に,LBプレート(+5%スクロース,−NaCl)に播種し,一晩培養する.
【7日目】
プレートに生えてきたコロニーに対し(図3D),コロニーPCRを行って目的の遺伝子が欠損しているかどうか確認する(Deletion check primerの位置を図2に示す).PCRの作業時に,同時に,コロニーのレプリカをLBプレート(−Amp)およびLBプレート(+Amp)にそれぞれとる.前者は,ストックのため,後者は,Amp感受性があることを確かめることで,確実にベクターが排除されていることを確認する.
増殖能などに関係の少ない遺伝子の欠損ならば,同一のプレート上の10個のコロニーに対してPCRを行えば,欠損株由来のコロニーが2個前後確認できる(その他は野生株由来)(図3E).
引用文献References
1) Ghatak, S., King, Z.A., Sastry, A., & Palsson, B.O. (2019) The y-ome defines the 35% of Escherichia coli genes that lack experimental evidence of function. Nucleic Acids Res., 47, 2446–2454.
2) Miller, J.H. (1992) in A short course in bacterial genetics: a laboratory manual and handbook for Escherichia coli and related bacteria, pp. 263–305, Cold Spring Harbor Laboratory Press.
3) Datsenko, K.A. & Wanner, B.L. (2000) One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. USA, 97, 6640–6645.
4) Li, Y. & Peng, N. (2019) Endogenous CRISPR-Cas system-based genome editing and antimicrobials: review and prospects. Front. Microbiol., 10, 2471.
5) Sekiya, K., Ohishi, M., Ogino, T., Tamano, K., Sasakawa, C., & Abe, A. (2001) Supermolecular structure of the enteropathogenic Escherichia coli type III secretion system and its direct interaction with the EspA-sheath-like structure. Proc. Natl. Acad. Sci. USA, 98, 11638–11643.
6) Simon, R., Priefer, U., & Puhler, A. (1983) A broad host range mobilization system for in vivo genetic engineering: transposon mutagenesis in gram negative bacteria. Nat. Biotechnol., 1, 784–791.
7) Miller, V.L. & Mekalanos, J.J. (1988) A novel suicide vector and its use in construction of insertion mutations: osmoregulation of outer membrane proteins and virulence determinants in Vibrio cholerae requires toxR. J. Bacteriol., 170, 2575–2583.
8) Nash, H.A. (1996) in Escherichia coli and Salmonella: cellular and molecular biology (Neighardt, F.C. ed.), 2nd ed., Vol. 2, pp. 2363–2376, ASM Press.
9) Gay, P., Le Coq, D., Steinmetz, M., Berkelman, T., & Kado, C.I. (1985) Positive selection procedure for entrapment of insertion sequence elements in gram-negative bacteria. J. Bacteriol., 164, 918–921.
10) Wek, R.C., Sameshima, J.H., & Hatfield, G.W. (1987) Rho-dependent transcriptional polarity in the ilvGMEDA operon of wild-type Escherichia coli K12. J. Biol. Chem., 262, 15256–15261.