1)パーキンソン病の病理形成
パーキンソン病(Parkinson’s disease:PD)は,主に中脳黒質にあるドーパミン作動性ニューロンが変性・脱落することで起こる進行性の神経変性疾患で,手足の震えや筋肉のこわばり,動きづらさなどの運動症状を特徴とする2).1000人あたり1~1.5人が罹患する,アルツハイマー病(Alzheimer’s disease:AD)に次いで2番目に頻度の高い神経変性疾患で,40歳以上の中高年で発症率が高くなる加齢性の疾患でもある.病変部位の神経細胞内にはレヴィ小体と呼ばれる封入体が形成され,その主要構成成分はαシヌクレインタンパク質の凝集体である.このαシヌクレイン凝集体が毒性を持ち神経細胞死を惹起することでPD発症に関与しているとの説が現在有力であるが,凝集体形成の分子機構ははっきりわかっていない.αシヌクレイン凝集体が中脳以外の部位で形成された場合,レヴィ小体型認知症(dementia with Lewy body:DLB)や多系統萎縮症(multiple system atrophy:MSA)の原因となることも知られており,これらαシヌクレインが関与する疾患はシヌクレオパチーと総称されている.
2)PDとグルコシルセラミダーゼ遺伝子変異
近年の疫学的研究から,グルコシルセラミダーゼ(glucosylceramidase:GBA)遺伝子変異がPDの強いリスク因子であることが示された.GBAはスフィンゴ糖脂質分解酵素の一つでグルコシルセラミド(GlcCer)からグルコースを除去する.GBA遺伝子変異は,ゴーシェ病と呼ばれるリソソーム蓄積病の原因でもある3).ゴーシェ病は遺伝性の代謝異常症であり,GBAの活性低下によって末梢組織ではマクロファージに基質のグルコシルセラミド(GlcCer)が,中枢神経系ではGlcCerとそのリゾ体であるグルコシルスフィンゴシンが蓄積することで発症する.このゴーシェ病のうち神経症状のないI型の患者がPD様症状を呈し,ゴーシェ病患者の家系内で(発症しない)GBA変異ヘテロ保有者にPD患者が頻出することが報告された4).このため2009年に日本を含めた世界の16施設が共同参加した国際研究が実施され,PD患者と健常者それぞれ約5000名についてGBA遺伝子が解析された.その結果,GBA遺伝子変異キャリアは非キャリアと比較して5.4倍のオッズ比でPDにかかりやすく,GBA遺伝子変異は現在知られているなかで最も強いPDのリスク因子であることが明らかになった5, 6).ちなみにPDを発症している人の約7%が変異型GBAのキャリアであり,L44PやN370Sなど特定の変異が多くみられるものの,マイナーなものを含めると30種以上の変異型が報告されている.またGBA遺伝子変異によってDLBの発症率も9倍程度上昇することなどから,GBA遺伝子変異はシヌクレオパチー全般のリスクファクターと認識されている7).
GBA遺伝子変異がどのような分子機序でPD病理形成に関与しているかは今のところ未解明である.現在そのメカニズムとして,変異型GBA自体が毒性を発揮するというgain of toxic function説と,GBAの活性低下,つまりスフィンゴ脂質代謝の変動が関係するというloss of function説の2通りが提唱されており議論が続いている8).gain of function説を支持する根拠としては,変異の大半がミスセンス変異であること,GBAがレヴィ小体に局在していることや,培養細胞系においてGBAタンパク質が直接αシヌクレイン凝集を誘導するという実験結果による.一方,loss of function説は,84GG, VA2+1GといったGBAを発現しないナンセンス変異型においても発症リスクの上昇がみられることがあげられる.ヒト型αシヌクレインを発現するキイロショウジョウバエPDモデルにおいても,GBA欠損でαシヌクレイン凝集体形成と運動行動異常が観察される9).コンズリトールBエポキシドなどを用いたGBA活性阻害がαシヌクレインの蓄積や凝集を誘導することが,培養細胞系やモデルマウスへの投与実験で実証されている10, 11).また培養細胞実験においてGBA活性とαシヌクレイン蓄積量に負の相関がみられている.PD患者由来のiPSニューロンを用いた実験では,GlcCer量やGlcCer/Cer比率とαシヌクレイン二量体/単量体比率が正の相関を示しており,GlcCer自体がαシヌクレイン凝集体の形成を誘導する可能性もある11).実際,合成リポソームを用いた実験ではGlcCerやGlcCerから脂肪酸が脱離したリゾ体のグルコシルスフィンゴシンが,αシヌクレインオリゴマーを安定化することで凝集体形成を促進することが示されている12).最近の研究ではマウス実験においてGBA遺伝子変異によるαシヌクレイン蓄積が酸性セラミダーゼの阻害で緩和されることが示されている13).これはαシヌクレイン蓄積にGlcCerではなくCerが関与することを示唆する報告で,この論文ではCerが分泌型オートファジーを誘導してαシヌクレイン蓄積を抑制するという機序が提案されている.さらに別の最近の研究でGBAにはGlcCer分解活性だけでなく,GlcCerのグルコースをコレステロールに転移しコレステリルグルコシドを生成する活性を持つことも報告された14, 15).GBA遺伝子変異が脂質プロファイルに及ぼす影響はより大きいと考えられ,どの脂質分子種がどのような機序でαシヌクレインの形成誘導を行っているのか,今後の研究の発展が待たれる分野となっている.
1)アルツハイマー病の病理形成
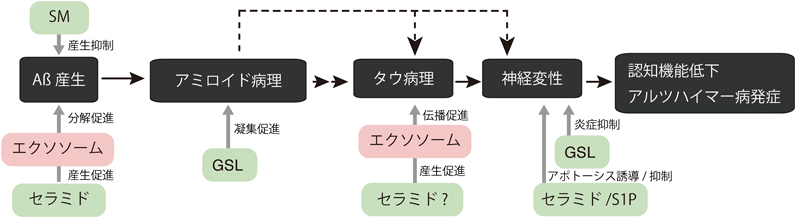
アルツハイマー病(AD)は,最も患者数の多い認知症で,進行性の神経変性疾患である.初期病理としてまずアミロイドβタンパク質(Aβ)のアミロイド凝集体が細胞外に沈着した老人斑が脳内に出現する(図2).続いて神経細胞内に異常リン酸化したタウタンパク質の凝集体である神経原線維変化(neurofibrillary tangle:NFT)が現れ,その後,神経変性が起こり認知機能が低下した結果,ADが発症する16).Aβは膜タンパク質のアミロイド前駆タンパク質(amyloid precursor protein:APP)が,βおよびγセクレターゼという二つの分解酵素によって切断を受けて産生される.家族性(遺伝性)ADでは,APPやセクレターゼの遺伝子変異によるAβ産生亢進が病因となるが,9割以上を占める孤発性ADでは明確なAβ沈着の原因はわかっていない.Aβ産生を標的とする薬剤の開発が世界中で進んでおり,介入時期を早めることで予防・治療効果を期待できると見込まれているものの,病理進行の機序については,アミロイド病理形成やタウ病理形成・拡散など不明な点が多く残っている.スフィンゴ脂質がこれらAD病理形成の複数のプロセスに関与することを示唆する研究が近年多数報告されている.
2)AD脳におけるスフィンゴ脂質代謝
AD患者脳においてスフィンゴ脂質濃度が変動することが,まだ少数ではあるが報告されている17).まず脳内Cer濃度は正常対照者と比較してAD患者の脳で増加していることが複数の研究グループから報告されており,その傾向も文献間で一致している18–21).ADに移行するリスクの高い軽度認知障害(mild cognitive impairment:MCI)対象者でも観察されており20),Cer増加がAD初期病理の原因となるか,もしくは結果として出現する可能性がある.またCerとともにSM濃度の報告も複数なされているがその傾向は一致しておらず,増加,減少または変動なしと文献間で相違がある18, 21–23).他の分子種に関しては,S1PがAD脳で増加すること,硫酸化GSLであるスルファチドの減少が報告されている18, 20, 24).最近では組織局所での変化を調べるためスフィンゴ脂質の質量分析イメージング技術の開発も進んでいる.現在のところADモデルマウスを用いた検証の段階であるが,Aβ沈着斑の近傍において特定の脂肪酸鎖長の脂肪酸を持つセラミドやGSLの変動がみられるなど興味深い観察結果が得られている25, 26).今後このように部位特異的な脂質変化が捉えられることで病理形成機序について議論が進むことが期待される.
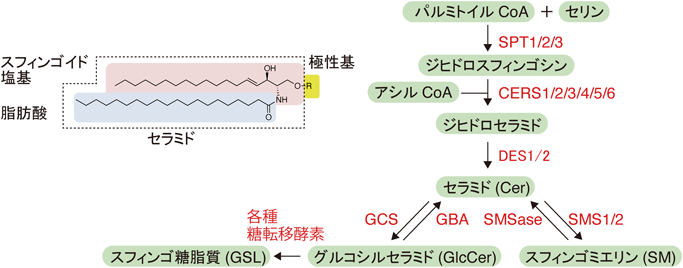
3)スフィンゴ脂質によるAβ産生制御
APPは主に神経細胞で発現する1型膜タンパクであり,翻訳後に細胞膜にリクルートされる.その後,アミロイド形成経路(amyloidogenic pathway)では,APPはエンドサイトーシスによって細胞内に取り込まれ,βおよびγセクレターゼの連続的な切断を受けAβが産生される.産生されたAβはまずエンドソーム内腔に放出され,エンドソームリサイクリングによって細胞外に分泌される.一方,非アミロイド形成経路(non-amyloidogenic pathway)では,αとγセクレターゼよる切断が行われ,Aβとは異なる毒性を持たないp3ペプチドが産生される.
スフィンゴ脂質の組成変動が,Aβ産生効率に影響を与える例が報告されている.APP遺伝子導入CHO細胞において,SPT阻害剤のミリオシン処理や不活性SPT導入によってスフィンゴ脂質全体を枯渇させると,Aβ産生量が増加する27).またこの報告とは矛盾するようであるが,同じくAPP遺伝子導入CHO細胞において,C6-Cer添加やSMase処理でCerを増加させた場合も,Aβ産生が促進される28).この論文によるとCer処理によってβセクレターゼの本体であるBACE1タンパク質の分子寿命が伸びることがAβ産生亢進に寄与するようである.またAβ1-42(重合性の高いAβ分子種)がSM分解酵素の中性SM分解酵素(nSMase)を賦活化し,さらにSMがγセクレターゼの活性を負に制御しているとの報告もある29).したがってAβ1-42濃度が高くなるとnSMaseが活性化しSMが減少する.その結果γセクレターゼが活性化し,Aβ1-42産生が促進されるという負のループが形成される可能性がある.実際にAβ1-42濃度が高いAD患者脳において,nSMase活性亢進やSM濃度低下が観察されている.また,βおよびγセクレターゼは細胞膜に存在するが,特にSM, GSLやコレステロールを多く含む脂質ミクロドメインと呼ばれる領域に局在することが知られている30, 31).SMによるγセクレターゼ活性抑制は,局所の脂質環境の酵素活性への影響が考えられる.またβセクレターゼについても活性本体のBACE1タンパク質を組み込んだ合成リポソームで膜脂質の影響を調べた実験によると,GlcCerやGalCerの存在でその活性が亢進されることが報告されている32).
4)スフィンゴ糖脂質によるアミロイド線維形成
GSL,特にシアル酸を含有するガングリオシドと呼ばれる酸性GSLにAβが結合することが以前から知られている.1995年に柳澤らは,初期AD病変を示す脳内にGM1ガングリオシド(GM1)と結合したAβ(GM1結合型Aβ, GAβ)が形成されていることを発見し報告した33).高齢になるとAD様の病変が現れる霊長類カニクイザルの脳内においても,加齢依存的にGM1の増加とGAβの出現が観察されている34).このGM1とAβの結合は人工脂質膜やリポソームを用いた実験でも確認されており,さらに原子間力顕微鏡解析からはAβはGM1分子1個ではなく複数が膜上で集積したクラスターに結合することがわかっている35).GM1ミセルを用いたNMR解析によると,Aβはもともとランダムコイル構造をとるが,GM1クラスターと結合したときにはアミノ酸配列の中央部分に二つのαヘリックス構造が形成される36).この際AβはGM1クラスターの親水性と疎水性の境界面に横たわるように位置しており,分子動力計算の結果も合わせて糖残基の根元側と脂質の頭部側が結合に重要であると考えられている37).また強弱の差はあるもののGM1以外のガングリオシド分子種のAβとの結合も確認されている38).
さらにADのアミロイド病理との関係において重要なのは,GAβがシード(種)となってAβのアミロイド線維化を誘導することである.GAβ存在下では自己重合が起こらない低濃度でもアミロイドが形成される39).NMR解析から,GM1クラスター上でAβが密になる条件において,C末端領域を介してAβ間の相互作用とβ構造への遷移が起こると考えられる40).加齢マウス脳シナプトソームでのGM1増加や,AD患者海馬でのGM1分布撹乱が観察されることから41, 42),これらの脳ではGAβ形成の基盤となるGM1クラスターが局所的に形成されやすい状態になっているのかもしれない.またGM2ガングリオシド分解酵素のヘキソサミニダーゼを欠損するサンドホフ病モデルマウスにおいて,ガングリオシド蓄積とともに神経細胞におけるGAβ形成やAβ蓄積が観察されている43).逆にヘキソサミニダーゼ活性のエンハンサーとして働く化合物をAPP遺伝子導入マウスへ経口投与すると,GAβが減少するとともに,脳神経細胞内のAβ蓄積の減少と認知行動の改善が認められる44).また最近Aβを発現するショウジョウバエのADモデルにおいても,GSL合成酵素遺伝子導入とシアル酸給餌によってガングリオシドを体内産生させるとAβ凝集体が増加することが報告されている45).これらの知見からGSLがAβ凝集に影響を与えることは明らかであり,今後この現象がAβ代謝やアミロイド病理形成にどのように関与しているか明らかになると期待される.加えてGM1を含むGSLは細胞膜上で脂質ミクロドメインに局在する.脂質ミクロドメインの他の構成脂質であるSMがGSLのクラスター化を牽引することも報告されており46),GSL以外のスフィンゴ脂質組成変動がGAβ依存性Aβ凝集という現象を通していかにAD病理に関与するかも興味深い.
5)エクソソーム産生制御を介したAD病理への関与
エクソソームは,さまざまな細胞から放出される直径が100 nm程度の細胞外小胞の一種である.multivesicular body(MVB)の内腔小胞がリサイクリングによって細胞外に放出されたものであり,多くの細胞外小胞のように細胞膜由来ではなくエンドソーム膜を由来とする.中枢神経系でも生理的に産生されており,マウス,ヒツジ,サルなどのモデル動物やヒト脳脊髄液(cerebrospinal fluid:CSF)中に存在することが確認されている47–49).また培養細胞においてニューロンやグリア細胞(アストロサイト,オリゴデンドロサイト,ミクログリア)からのエクソソーム放出が確認されている50–52).脳内エクソソームの役割については発生期のミエリン成熟や神経突起伸長などの生理的役割が報告されている一方で,PDやAD,プリオン病,筋萎縮性側索硬化症(amyotrophic lateral sclerosis:ALS)などさまざまな神経変性疾患の関連分子がエクソソームに含まれることが報告され,これら疾患分子の脳内代謝や伝播にエクソソームが関与する可能性も取り沙汰されている25).
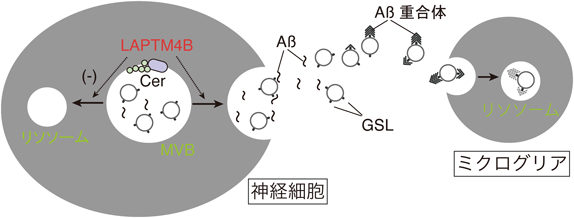
AβはAPP遺伝子導入マウス(APPマウス)やヒトのCSFや血液中のエクソソームに含まれることが報告されている.上述のようにAβはMVBを含むエンドソームでAPPから切り出され内腔側に放出される.このため,Aβとエクソソームは表面膜を介して相互作用すると考えられ,実際我々のグループでは以前,共鳴プラズモン共鳴法などを用いて,培養神経細胞(N2a細胞,マウス脳初代神経細胞)から調製したエクソソームの表面にAβが結合することを確認した53).またエクソソームの膜脂質を解析したところ,GM1をはじめとしたGSLが,由来細胞と比較して多量に含まれていることがわかった54).またエンドグリコシルセラミダーゼ処理でGSLの糖鎖を除去したエクソソームではAβ結合性が失われることから,Aβは表面GSL糖鎖を介して神経細胞由来エクソソームに結合すると考えられる.ちなみに培養ミクログリアやアストロサイト由来のエクソソームはGSL含量が低く,Aβ結合性は認められない.前項で述べたようなGSLクラスターがエクソソーム膜上にできてAβ結合の場が形成されると考えられる.実際,神経細胞由来エクソソームはAβ結合だけでなくAβアミロイド線維形成も促進する53).
また,放出されたエクソソームは主に,脳内の貪食細胞であるミクログリアに取り込まれる55).エクソソームに結合したAβも一緒にミクログリアに貪食され,リソソームで分解される.APPマウスの脳へ培養神経細胞由来のエクソソームを注入すると,脳内Aβとの結合,ミクログリアへの移行が観察される54).また浸透圧ミニポンプを用いてエクソソームを2週間持続投与すると,脳内Aβが減少し,アミロイド沈着が低減する.神経細胞由来のエクソソームは,GSLでAβを捕捉しミクログリアへ送達することで,Aβクリアランスを促進すると考えられる(図3).一方で,加齢やAD発症に伴ってミクログリアの活性が低下するといった報告も多く,この場合,エクソソーム結合Aβは細胞外空間にとどまり老人斑形成の核となったり,後述するタウ病理のように神経細胞間のAβ凝集体伝播に関与する可能性も想定される.
Aβ結合にGSLが関与するだけでなく,エクソソームの産生にもスフィンゴ脂質が関与することが明らかになっている.神経細胞由来エクソソームがAβクリアランスを補助することが明らかになったので,次に我々は,エクソソーム産生を亢進させる方法はないかと考えた.2008年にSimonsらのグループから,Cer依存的にエクソソームが産生されることが報告されている56).エンドソーム膜のSMがnSMaseに分解されてCerが増加すると,膜が物理的性質の変化によって内腔側に陥入し内腔小胞が形成される.こうしてできたMVBによって結果的にエクソソームが増加することになる.セラミド依存性(nSMase依存性)とは別に,MVBの内腔小胞形成にはESCRT(endosomal sorting complex required for transport)複合体が関与する系が知られているが,両者は独立して働くとされている56).
これらの知見から,我々のグループではエクソソーム産生亢進のためにCerが利用できるのではないかと考え,神経細胞株SH-SY5Yの培養液中にCerを直接添加する実験を行った.その結果,この外部添加Cerによってもエクソソームの増加が認められた57).また増加したエクソソームもGSLを含有しておりAβ結合性を維持していた.トランスウェル培養系において,インサート内にAPP遺伝子導入SH-SY5Y細胞,下部ウェルにミクログリア細胞株BV-2を播種して共培養すると,SH-SY5Y細胞から分泌されたAβのBV-2細胞への取り込みが観察される.この系の培養液中にCerを添加するとBV-2のAβ取り込みが促進され,培養液のAβ濃度が減少する.Cer外部処理によってAβ分解促進が起こったと考えられる.また最近の我々のAPPマウスを用いた研究では,植物由来Cerを2週間経口投与すると脳内Aβ濃度が減少するという結果も得られている57).植物由来Cerは,哺乳動物Cerと構造が若干異なりスフィンゴイド塩基の8位部分に二重結合が一つ多いが,同様にエクソソーム産生促進効果を示す.植物Cer投与マウスの脳組織と血清からエクソソームを調製して解析すると,総粒子数には差が認められないものの,神経細胞マーカータンパク質NCAM-1とL1CAMが増加した.またエクソソーム含有Aβ量もCer投与群で増加していた.体内において経口投与セラミドでエクソソーム依存性Aβ除去系が賦活化している可能性が示唆される結果である.
また最近我々は,外部添加Cer依存的なエクソソーム産生誘導機構についてSH-SY5Y細胞を用いて調べた(未発表データ).外部から添加したCerは,当初nSMase依存産生系と同様に,ESCRT依存産生系と独立して作用すると考えたが,ESCRT構成タンパク質のHSGやTsg101のノックダウンによって外部Cer処理の効果が阻害されたことから,ESCRT依存系と関連することが示唆された.さらに異なる鎖長の脂肪酸を持つCerで効果を比較したところ,C16やC18の炭素鎖長を持つ長鎖脂肪酸結合Cerで最もエクソソーム産生誘導作用が強く,C2やC6の短鎖脂肪酸やC24の極長鎖脂肪酸を持つCerには効果がなかった.またSMやGlcCer処理でもエクソソーム量に変化はなかった.長鎖脂肪酸結合Cerは処理後にエンドサイトーシスによって取り込まれリソソームに蓄積することから,我々は,リソソームや後期エンドソームに局在するタンパク質でセラミドと相互作用することが報告されているlysosomal-associated transmembrane protein 4B(LAPTM4B)58, 59)に注目し実験を行った.lipid–protein overlay assayでセルロース膜上にスポットした脂質とLAPTM4Bタンパク質との結合を調べたところ,エクソソーム産生誘導作用の強い長鎖脂肪酸結合Cerと強い結合がみられた.SMやGlcCerとの結合は認められなかった.そしてsiRNAでLAPTM4B遺伝子をノックダウンした細胞では外部Cer処理によるエクソソーム増加がほぼ完全に阻害されることから,外部添加CerはLAPTM4Bを介してエクソソーム産生を誘導すると考えられた.また酸性セラミダーゼ阻害でもLAPTM4B依存性のエクソソーム産生が誘導されることが明らかになった.酸性セラミダーゼは,リソソーム内でCerをスフィンゴイド塩基と脂肪酸に分解する酵素で,活性阻害するとCerが蓄積することが知られており,この酵素の遺伝子欠損はファーバー病というリソソーム蓄積病の原因となっている60).またCer処理後,GFP標識CD63(MVBマーカータンパク質)発現細胞を観察すると,MVBとリソソームの共局在率が減少し,リサイクリングエンドソームのマーカータンパク質であるRab11との共局在率が上昇した.この共局在率の変化は酸性セラミダーゼ阻害でも同様に起こった.
さらにCerはエクソソームに含まれて細胞外放出されることも知られている61).我々の使用した培養細胞系では,酸性セラミダーゼ阻害下でも細胞内Cer量は変化しなかったが,LAPTM4Bノックダウンでエクソソーム産生を抑制すると細胞内(リソソーム内)におけるCer量が顕著に増加した.これらの結果は,LAPTM4Bによってエンドソーム・リソソーム内のCer蓄積が感知され,MVB輸送方向の転換によってエクソソーム放出が誘導されることが示唆される.前述したようにMCIや初期AD患者脳でCer増加が認められることから,この病理過程にエクソソーム産生が関与する可能性も考えられ,今後研究を進めていきたい.
またエクソソームはタウ病理の脳内での拡散に関与する可能性があることが報告されている62).タウ病理のNFTはまず旧内皮質の神経細胞に現れ,その後神経回路の投射先である海馬の神経細胞でも観察されるようになる.池津らのグループでは,このタウ病理の伝播を短期間で再現できるモデルマウスを作製し実験を行ったところ,ミクログリアを欠損させた場合に,タウ病理の伝播が抑制されることを明らかにした.また細胞培養系において,神経細胞で形成されたタウ凝集体を取り込んだミクログリアは,タウを含有したエクソソームを放出すること,さらにこのエクソソームが別の神経細胞に取り込まれ,その細胞内でタウ凝集体形成が誘導される.これらの結果からは,ミクログリアによるタウ凝集体の取り込みと,エクソソームに封入されたかたちでの再放出,そのエクソソームの神経細胞へのターゲッティングがタウ病理拡散に関与することが示唆される.またマウス実験では,nSMase阻害剤を投与するとタウ病理伝播が抑制されことから,この系のエクソソーム産生過程,もしくは別のステップにSMやセラミドが関与している可能性も考えられ,現在検討されている.またエクソソームにはαシヌクレインも含まれその脳内拡散に関与することを示唆する研究も報告されている63).GBAによる生成物の一つはCerでありPD病理形成においてもエクソソームが関与するか今後の研究の進展が興味深い.