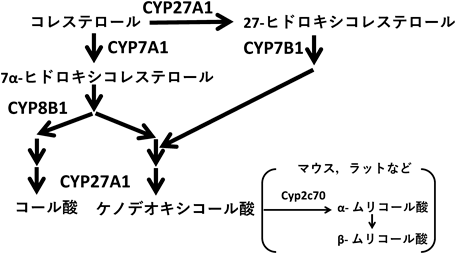
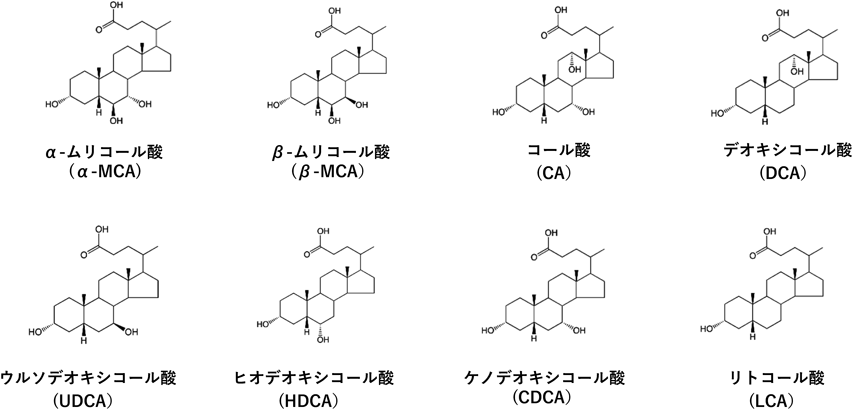
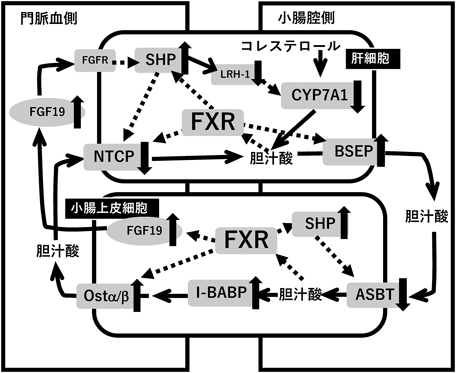
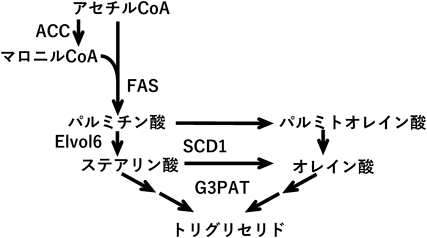
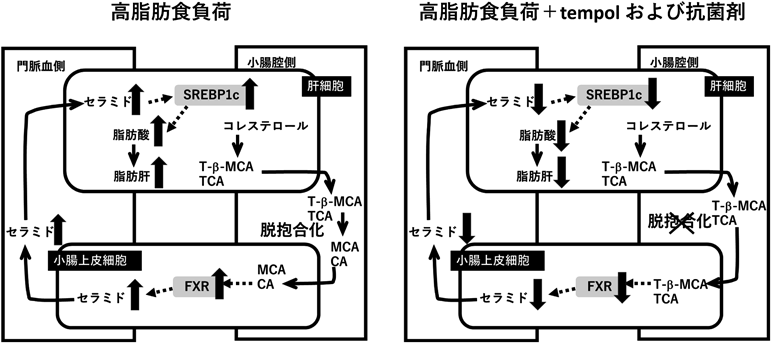
胆汁酸による脂肪合成系の制御Regulation of lipogenesis by bile acids
富山大学学術研究部薬学・和漢系Laboratory of Nutritional Biochemistry, Institute of Natural Medicine, University of Toyama ◇ 〒930–0194 富山市杉谷2630 ◇ 2630 Sugitani, Toyama 930–0194, Japan
発行日:2020年10月25日Published: October 25, 2020