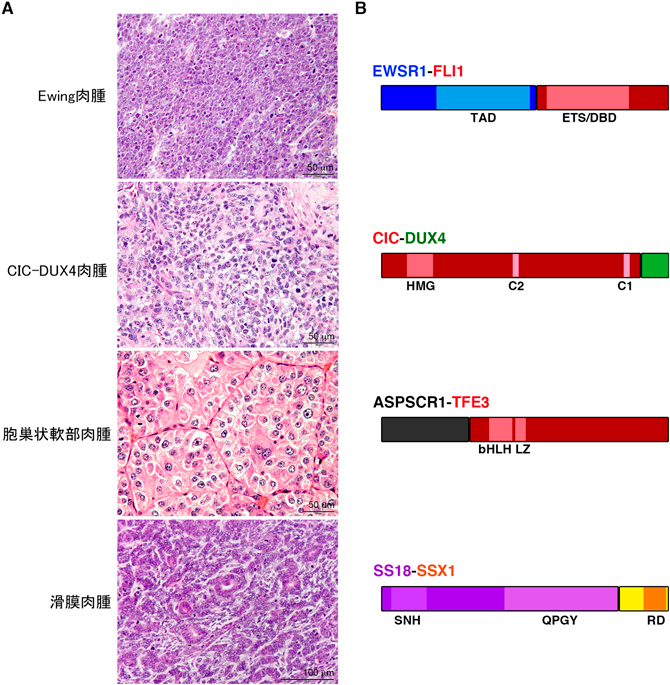
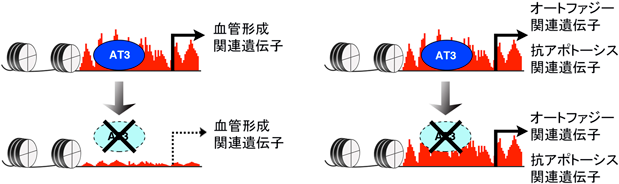
融合遺伝子特異的な骨軟部肉腫の誘導Induction of fusion gene-associated bone and soft tissue sarcoma
公益財団法人がん研究会 がん研究所 発がん研究部Division of Carcinogenesis, the Cancer Institute, Japanese Foundation for Cancer Research ◇ 〒135–8550 東京都江東区有明3–8–31 ◇ 3–8–31 Ariake, Koto-ku, Tokyo 135–8550, Japan
発行日:2020年12月25日Published: December 25, 2020