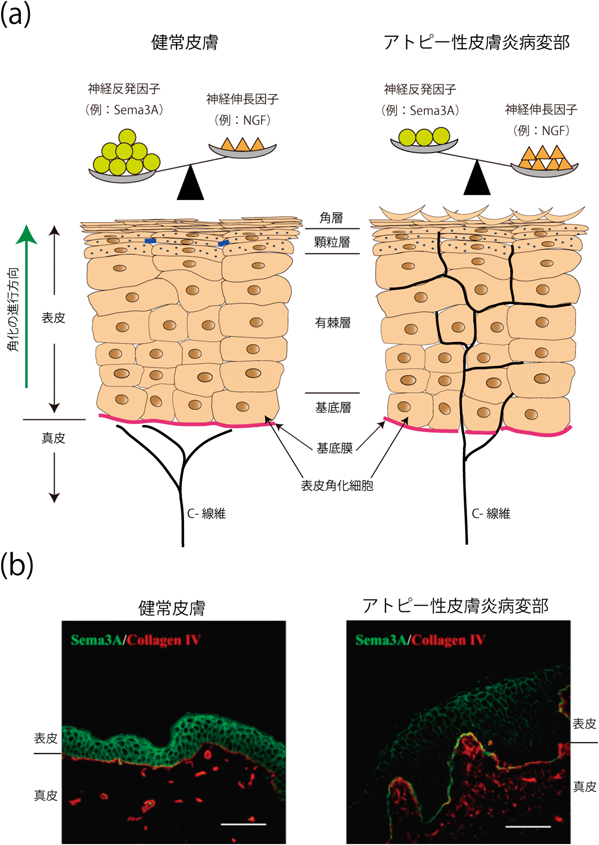
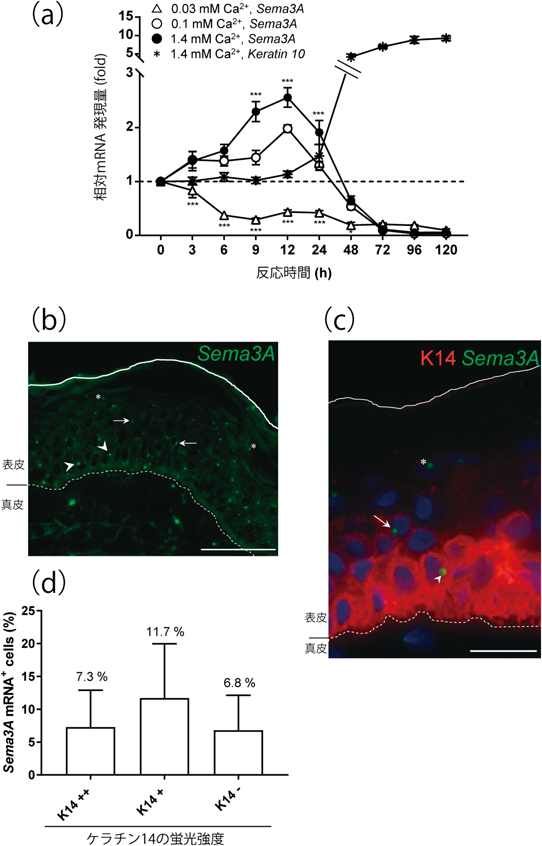
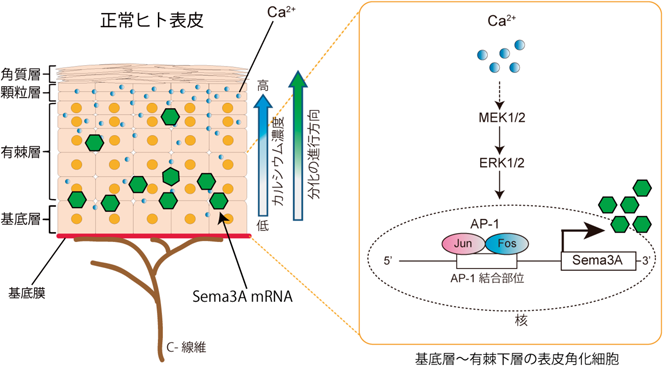
ヒト表皮角化細胞におけるセマフォリン3Aの発現制御機構Regulatory mechanisms of semaphorin 3A expression in human keratinocytes
順天堂大学大学院医学研究科環境医学研究所・順天堂かゆみ研究センター(JIRC)Juntendo Itch Research Center (JIRC), Institute for Environmental and Gender-Specific Medicine, Juntendo University Graduate School of Medicine ◇ 〒279–0021 千葉県浦安市富岡2–1–1 ◇ 2–1–1 Tomioka, Urayasu, Chiba 279–0021, Japan
発行日:2020年12月25日Published: December 25, 2020