わが国では,食の欧米化や運動不足などに伴った過栄養による生活習慣病の発症増加が社会問題となっている.生活習慣病の発症リスクを高める要因の一つとして,脂肪肝がある.脂肪肝の症例は肥満を伴っているものが多く,飲酒以外の脂肪肝形成の主要因として考えられているのは,過栄養によって起こる糖・脂質代謝の調節異常である.他方,肥満を伴わない脂肪肝も一定数認められており1, 2),その原因の一つは,必要な栄養素を十分に摂取できていない低栄養である.過栄養と低栄養は正反対の栄養状態であるが,いずれの栄養状態も栄養素の必要量と摂取量のアンバランスから生じて代謝異常を引き起こし,脂肪肝を形成しうる.脂肪肝は,単に生活習慣病のリスクファクターであるだけでなく,肝炎,肝硬変へと病態が進展して,肝不全を招く恐れがある.したがって,食習慣に起因した不適切な栄養状態に応答して肝臓に過剰な脂質が蓄積するメカニズムを解明することは,生活習慣病の予防法や治療法の開発に役立つのみならず,健康寿命延伸のためにも重要な課題である.
過栄養による肥満や2型糖尿病を伴った脂肪肝の発症機序については,モデル動物を用いた研究成果が数多く報告されており,インスリンが十分量あるにもかかわらずその作用が発揮されない「インスリン抵抗性」との関連が指摘されている.その一方で,低栄養による脂肪肝の発症機序に関する研究報告は少ない.低栄養のうち,摂取するタンパク質の量が不足してタンパク質栄養状態が悪化すると,ヒト,サル,ラットなどで脂肪肝になることは,1950年代から報告されていた3–5).そして,長年,その原因は,肝臓からの中性脂肪(triglyceride:TG)の放出の低下と考えられていた6, 7).しかし,近年の研究成果から,タンパク質栄養状態の悪化による脂肪肝の形成は,ホルモンの変化,肝臓内での代謝の変化,肝臓以外の臓器での代謝の変化などが互いに影響しあった結果起こることがわかってきた.
本稿では,摂取するタンパク質の「量」の不足や「質」の低下によって肝臓に過剰な脂肪蓄積が起こるメカニズムについて,近年我々が推し進めてきた研究内容を中心に概説したい.
成長期の動物のタンパク質栄養状態が悪化すると,さまざまなホルモンの血中濃度が変化し,これにより代謝に変化が生じることが知られている.そこで我々は,肝脂肪蓄積に何らかのホルモンが関与している可能性があるのではないかと考え,検討を行った.具体的には,タンパク質栄養状態の悪化によって血中濃度が変化するホルモンについて,ホルモンの遺伝子欠損あるいはホルモンの投与が肝臓脂肪蓄積に及ぼす影響を解析した.
1)インスリン
低タンパク質食を摂取しているラットでは,インスリン分泌が低下することが報告されているが,この低下は二つの機構で生じる.一つは比較的長期間のタンパク質欠乏で生じる,膵島の萎縮によるインスリン分泌の低下である8).この場合,膵臓のインスリン分泌細胞の量が低下しているため,グルコースにより誘導されるインスリン分泌が低下する.もう一つは,タンパク質含量が少ない飼料のインスリン分泌促進作用が小さいという機構である.インスリンは基本的には食餌中の糖によって分泌が誘導されるが,タンパク質・アミノ酸にもインスリン分泌を促進する作用がある.タンパク質含量が少ない飼料ではこの活性が低いため,摂食時のインスリン分泌が低くなる.我々は,タンパク質含量が20%の対照食と比較して5%の低タンパク質食では,ラットの摂食時の血中インスリン濃度が顕著に低くなることを示している9).さらに,分岐鎖アミノ酸含量が1種類でも少なくなると摂食時のインスリン分泌が低下すること,低タンパク質食に3種の分岐鎖アミノ酸を対照食レベルまで添加するとインスリン分泌が回復することを報告し,インスリン分泌にはタンパク質中の分岐鎖アミノ酸含量が重要であることも明らかにした10).
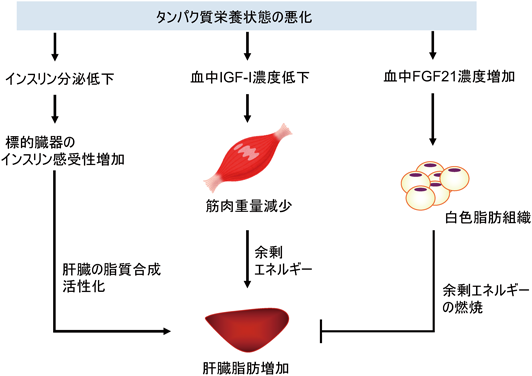
詳細は「3. 肝臓における調節」で述べるが,タンパク質欠乏時にはインスリン分泌が低下する一方,標的臓器のインスリン感受性は上昇しており,肝臓でのインスリン作用の増加が脂肪肝の原因となる可能性が考えられた.そこで,インスリン分泌の低下がインスリン感受性の増加を介して脂肪肝の原因となる可能性を検討する目的で,低タンパク質食摂取ラットにインスリンを投与して血中濃度の低下を抑制し,脂肪肝への影響を解析する実験を行った.タンパク質を15%含む対照食あるいは5%含む低タンパク質食を与えたWistar系雄ラットに,持続型(中間型)ヒトインスリンを毎日摂食開始前と開始3時間後の2回投与して,7日間飼育した.その結果,低タンパク質食摂取による血中インスリン濃度の低下を抑制することができた.このとき,インスリン非投与群では低タンパク質食摂取による肝臓中性脂肪の約3倍の増加が確認されたが,インスリン投与群でもこの増加は抑制されなかった.したがって,タンパク質欠乏時に生じるインスリン分泌の低下は,脂肪肝の直接の要因ではないことが明らかになった11).インスリン分泌低下が脂肪肝形成の主要因ではないことが示されたが,インスリン作用の増加が脂肪肝形成に寄与する可能性は高いと考えられる.
2)insulin-like growth factor(IGF)-I
成長期の動物では,タンパク質栄養状態の悪化によって成長が抑制される.この成長抑制には,肝臓で合成されるIGF-Iが重要な役割を果たしていることが知られている.IGF-Iは成長ホルモンによって合成が促進されるタンパク質同化活性を持つホルモンで,成長ホルモンによる成長促進作用を媒介し,実質的に成長を担うホルモンである12).IGF-Iの合成および血中濃度はタンパク質栄養状態をよく反映するのが特徴で,摂取タンパク質の量の低下のみならず,アミノ酸バランスの悪化にも鋭敏に応答して低下する.1種類の必須アミノ酸欠乏でも血中濃度が大きく低下し,IGF-Iの血中濃度と成長期の体重増加は非常によく相関することを我々は報告している13).低タンパク質食摂取時の成長低下は,筋重量の低下や筋肉におけるタンパク質代謝活性の低下を伴っている.この低下は筋肉でのエネルギー消費の減少を引き起こし,余剰となったエネルギーが肝臓に脂肪として蓄積する可能性が考えられた.そこで,この機構を介して,IGF-Iの血中濃度の低下が脂肪肝形成を引き起こす可能性について検討を行った.
成長期(7週齢)のC57BL/6マウスにタンパク質を15%含む対照食あるいは3%含む低タンパク質食を10日間与えた.各食餌群の半数のマウスに浸透圧ポンプでrecombinant human IGF-Iを投与することで,低タンパク質食摂取による血中IGF-I濃度の低下を抑制することができた.さらに,IGF-Iの投与により低タンパク質食摂取による体重低下および筋肉重量の低下を一部抑制することができた.しかし,タンパク質欠乏による肝臓脂肪の増加は,IGF-I投与により抑制されなかった.この結果から,タンパク質摂取量の血中IGF-I濃度の低下は体重や筋肉量の低下を引き起こすが,脂肪肝を引き起こす主要因とはならないことが明らかになった.詳細は後述するが,タンパク質欠乏時に筋肉量の低下によって生じる余剰のエネルギーは,脂肪肝形成に寄与していると考えられるが,脂肪肝形成の主要因ではないと考えられる.
3)fibroblast growth factor(FGF)21
我々はタンパク質欠乏に応答して合成が変化する分子をトランスクリプトーム分析により探索し,低タンパク質食を摂取するラットやマウスの肝臓で,FGF21の遺伝子発現が顕著に増加することを見いだした14).このとき,FGF21の血中濃度も大幅に増加していた.FGF21の増加は,二つの面でタンパク質欠乏と関連する可能性がある.一つは,FGF21の増加がIGF-I合成と成長の低下を引き起こす可能性で,FGF21トランスジェニックマウスでこれらの現象が生じることが報告されている15).もう一つはFGF21の脂質代謝調節作用で,これが脂肪肝形成に何らかの影響を及ぼす可能性が想定された16).そこで,タンパク質低欠乏時の血中FGF21濃度の増加が成長低下および肝臓脂肪増加に及ぼす影響を,FGF21遺伝子欠損マウスを用いて検討した.
成長期(4~5週齢)のFGF21欠損マウスおよび野生型マウスに,タンパク質を15%含む対照食あるいは5%含む低タンパク質食を11日間与えた.野生型マウスではタンパク質欠乏によりIGF-Iの肝臓mRNA量と血中濃度が減少し,体重増加が抑制された.また,肝臓FGF21 mRNA量および血中FGF21濃度はいずれも約10倍増加した.一方,FGF21の欠損はIGF-Iおよび成長の低下に影響を与えなかったことから,FGF21の増加はタンパク質栄養状態の悪化によるIGF-I低下や成長低下の主要因ではないことが示された.しかし,タンパク質欠乏時の成長低下がFGF21欠損で抑制されるという結果が他グループから報告されていることから17),FGF21の寄与は実験期間や食餌条件によって変動するものと考えられた.また,FGF21欠損マウスではタンパク質欠乏時の肝臓脂肪蓄積量がさらに増加するという結果を得た.この結果は,タンパク質欠乏により増加するFGF21は,脂肪肝を抑制する働きを持つことを示していた.FGF21は脂肪組織での熱産生を誘導するため,エネルギーを燃焼させることによって肝臓への脂肪蓄積を抑制しているものと考えられる18).この結果から,タンパク質欠乏時にはFGF21が増加してエネルギー消費が増加しているが,消費しきれない分が肝臓に脂肪として蓄積していることがわかる.タンパク質栄養状態が悪化すると,体内で消費しきれないエネルギーの肝臓への蓄積が脂肪肝形成に寄与することを示す結果であると考えている.
以上述べてきたように,タンパク質栄養状態の悪化によって,同化作用のあるインスリンやIGF-Iの合成・分泌が低下する.糖・脂質代謝を制御する作用が強いインスリンでは感受性が増加してホルモン濃度低下の糖代謝への目立った影響はみられなくなるが,タンパク質代謝制御活性が強いIGF-Iは活性が低下し,体内で余剰のエネルギーを生じる原因となる.この余剰エネルギーの肝臓への移行は,主要因ではないものの,脂肪肝形成に寄与するものと考えられた.一方で,エネルギーの燃焼を促進するFGF21の活性が増加し,肝臓に脂肪が蓄積するのを抑制するシステムが作動していることが明らかになった.栄養状態が悪い動物の体内で余剰のエネルギーを消費する仕組みが働いていることは意外に感じられるが,重要な適応現象であると考えられる.
次に,タンパク質栄養状態の悪化に応じて変動するホルモンなどの調節を受けて,肝臓内でどのような代謝変化が起こって,過剰な脂肪蓄積が起こるのかを分子レベルで検討した研究結果を紹介したい.
1)インスリンシグナルによる調節(図2)
脂肪肝とインスリン作用の関わりは,過栄養による肥満や2型糖尿病のモデル動物を用いた研究成果によって明らかになってきている.
インスリンがその作用を発現するのには,インスリンの細胞膜上にある受容体への結合を端緒とした細胞内シグナルの活性化が必要である.インスリンが受容体に結合し,受容体チロシンキナーゼが活性化されると,これがインスリン受容体基質(insulin receptor substrate:IRS)をチロシンリン酸化する.引き続き,チロシンリン酸化されたIRSにホスファチジルイノシトール3-キナーゼ(PI3キナーゼ)の調節サブユニットが結合することで,PI3キナーゼ経路やラパマイシン標的タンパク質複合体1(mechanistic target of rapamycin complex 1:mTORC1)経路が活性化され,インスリン作用が発現する19).
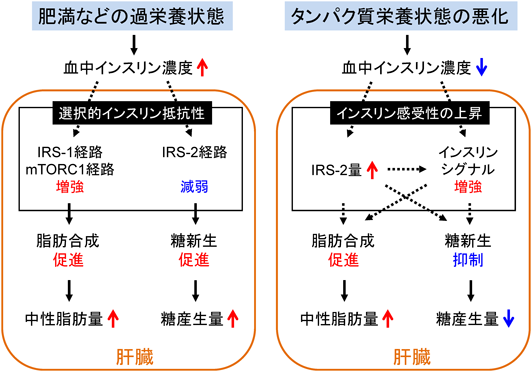
インスリンは,肝臓において,糖産生を抑制し,脂質合成を促進する.肝特異的インスリン受容体欠損マウスでは,肝臓におけるインスリン作用を完全に失った「全体的インスリン抵抗性(total insulin resistance)」状態にある20).このマウスでは,インスリンによる糖新生の抑制も,脂肪合成の促進も起こらず,脂肪肝の形成も起こらない21, 22).これに対して,肥満や2型糖尿病のモデル動物の場合では,インスリンによる糖新生抑制作用が低下して高血糖が惹起される一方で,インスリンの脂質合成促進作用は亢進して脂肪肝,高脂血症を併発する23, 24).このように,糖代謝においてインスリン抵抗性を呈し,脂質代謝においてインスリン感受性が上昇している状態は,「選択的インスリン抵抗性(selective insulin resistance)」と称されている20).そして,このような状態は,肥満や2型糖尿病を伴った過栄養による脂肪肝の発症メカニズムの一つとして考えられている.選択的インスリン抵抗性状態で脂質代謝のインスリン感受性が上昇する分子機構として,IRSの2種のサブタイプであるIRS-1とIRS-2の肝臓内での発現領域が異なるため,領域特異的なインスリンシグナルの調節によって起こる脂質合成の促進25)や,栄養シグナルによって活性化したmTORC1を介した脂質合成の促進26)などが報告されている.
他方,低栄養によって発症する脂肪肝とインスリン作用の関係はよくわかっていなかった.そこで,我々は,低タンパク質食を摂取させて肝TG量が顕著に増加したラットを用いて,タンパク質栄養状態の悪化が血糖値や血中インスリン濃度の変化に与える影響を調べた.その結果,このラットでは,膵臓からのインスリン分泌が抑えられる一方で,インスリン感受性は上昇し,血糖値が正常に維持されていた9, 27).また,肝臓におけるインスリンの細胞内シグナル因子の変化についても検討した.その結果,低タンパク質食摂取ラットの肝臓では,対照食摂取ラットの肝臓と比べて,IRS-2の量が増加し,摂食に応答したIRS-2のチロシンリン酸化量も増加することがわかった9).さらに,低タンパク質食摂取ラットの肝臓から単離した肝細胞を用いて,インスリンシグナルの変動を検討したところ,対照食摂取ラットの肝細胞と比べて,インスリン刺激に応答したインスリン受容体のチロシンリン酸化,Aktのセリン・トレオニンリン酸化が増強していた28).したがって,タンパク質栄養状態が悪化した場合には,肝臓におけるインスリンシグナルが増強すると同時に,脂肪肝になることがわかった.
以上の結果から,低タンパク質食を摂取した際には,インスリンによる脂質合成促進活性がインスリンシグナルの増強を介して上昇し,肝TG量の増加が起こるのではないかと考えられた.そこで,これを明らかにするために,低タンパク質食摂取ラット由来の肝細胞を用いて検討した.その結果,脂質合成調節酵素であるアセチルCoAカルボキシラーゼ1(acetyl-CoA carboxylase:ACC1)や脂肪酸合成酵素のmRNA量は,対照食摂取ラットと低タンパク質食摂取ラットのいずれの肝細胞においても,インスリン刺激依存的に増加した.しかし,これらのmRNA量は,インスリン刺激の有無にかかわらず,両群間で差は観察されなかった.単位時間あたりのTG合成量は,対照食摂取ラットの肝細胞に比べて,低タンパク質食摂取ラットの肝細胞の方が多かったが,両群ともにインスリン刺激によって増加しなかった28).したがって,タンパク質栄養状態が悪化した際の肝臓では,インスリン刺激の有無にかかわらず,脂質合成が促進し,TG量が増加することがわかった.
以上の結果をまとめると,低タンパク質食摂食によって起こるIRS-2量の増加やインスリンシグナルの増強が,インスリン刺激の有無にかかわらず,肝臓の脂質合成量を増加させていると考えられる.また,低タンパク質食摂取ラットの肝臓では,肥満や2型糖尿病の「選択的インスリン抵抗性」とは異なり,脂質合成が促進しているのと同時に糖新生が抑えられていた29).このことから,IRS-2量の増加やインスリンシグナルの増強は糖新生の抑制に対しても重要な役割を果たす可能性が考えられる.このように,タンパク質栄養状態の悪化によって起こる肝インスリンシグナルの増強の生理的意義については解明すべき点が多く残っており,さらなる研究成果が期待される.
2)翻訳調節因子を介した調節(図3)
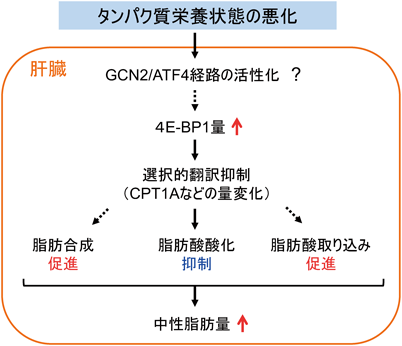
我々は,タンパク質栄養状態の悪化が肝臓のインスリンシグナルに及ぼす影響を検討する過程で,低タンパク質食を14日間与えたラットの肝臓で翻訳抑制因子4E-BP1(eukaryotic translation initiation factor 4E-binding protein 1)の量が増加することを見いだした9).
一般的に,4E-BP1は翻訳開始因子eIF4E(eukaryotic translation initiation factor 4E)に結合することで,翻訳を阻害する30, 31).4E-BP1とeIF4Eの結合は,4E-BP1のリン酸化によって調節されている.活性化されたmTORC1によって4E-BP1がリン酸化されると,4E-BP1がeIF4Eから解離し,翻訳が促進する32).このように,mTORC1-4E-BP1経路はタンパク質合成を翻訳開始段階で調節することが広く知られている.したがって,当初,タンパク質栄養状態の悪化に応答した肝4E-BP1量の増加は,肝臓におけるタンパク質合成を抑制するために起こると考えていた.
ところが,昨今,mTORC1経路はタンパク質合成の調節だけでなく,脂質代謝の調節にも深く関与していることを示した研究成果が多数報告されている.特に肝臓において,mTORC1の活性化が,脂質合成調節酵素の遺伝子発現に重要な転写因子であるSREBP-1c(sterol regulatory element-binding protein-1c)の発現および活性化を誘導し,脂質合成を促進することが報告されている33).これに加えて,mTORC1の基質である4E-BP1自身に関しても,脂質代謝への関与がいくつか報告されている.4E-BP1欠損マウスでは,野生型に比べて,エネルギー消費が亢進し,白色脂肪組織量が減少していた34).また,ショウジョウバエの4E-BP欠失変異体では,野生型に比べて,飢餓に応答した脂肪分解が促進していた35).さらに,我々の研究においても,タンパク質栄養状態の悪化に応答して起こる4E-BP1量の増加は,ラットに低タンパク質食を1日与えただけで観察され,肝TG量の増加はその後に顕著となることがわかり,4E-BP1量の増加がTG量増加を引き起こしている可能性が考えられた36).
そこで,低タンパク質食を摂取したラットの肝臓で起こる4E-BP1量の増加が,肝TG量の増加に直接的に関与しているか調べるために,肝臓の4E-BP1をノックダウンしたラットに低タンパク質食を与えた際の肝TG量の変化を検討した.その結果,肝4E-BP1をノックダウンしたラットでは,正常ラットに比べて,低タンパク質食によって起こる肝TG量の増加が抑えられた36).したがって,タンパク質栄養状態が悪化した際に起こる肝TG量の増加には,肝4E-BP1量の増加が必要であることが明らかとなった.また,この4E-BP1を介した肝TG量の調節機構に,どのような脂質代謝調節因子が関わっているのか調べた.その結果,低タンパク質食によって,脂肪酸合成に関わるACC1と脂肪酸取り込みに関わるCD36(cluster of differentiation 36)のタンパク質量は増加し,脂肪分解に関わるATGL(adipose triglyceride lipase)のタンパク質量は低下したが,これらの変化は4E-BP1ノックダウンの影響を受けなかった.他方,脂肪酸酸化の調節酵素であるCPT1A(carnitine palmitoyltransferase 1A)量は,低タンパク質食と4E-BP1ノックダウンの両方の影響を受けて変化し,低タンパク質食を与えた肝4E-BP1ノックダウンラットの肝臓で増加していた.そして,この増加はmRNAレベルではなく,タンパク質レベルで起こっていた36).これらの結果から,タンパク質栄養状態の悪化に応答して肝臓で脂肪酸酸化が抑制されること,そのメカニズムには4E-BP1による選択的な翻訳調節が関与していることが示唆された.
一連の知見は,肝臓において,4E-BP1がタンパク質・アミノ酸の栄養シグナルと脂質代謝とを結びつける重要な因子であることを示している.低タンパク質食による4E-BP1量の増加はどのような分子メカニズムで起こるのか,そのメカニズムにタンパク質・アミノ酸欠乏によって活性化されるGCN2(general control nonderepressible 2)−ATF4(activating transcription factor 4)経路37)が関与しているのか,CPT1A以外にどのような脂質代謝調節因子が4E-BP1を介した翻訳調節によって制御されているのかは,今後の課題である.
「3. 肝臓における調節」で述べたように,低タンパク質食摂取により肝臓ではインスリンシグナル伝達分子や翻訳調節因子4E-BP1に変化が生じ,脂肪肝発症を引き起こすことが明らかになった.これに加え,肝臓以外の臓器で生じる変化も肝臓への脂肪蓄積に寄与する.ここでは,特にエネルギー消費の多い筋肉が脂肪肝に影響を及ぼすことを示す研究結果を紹介したい.
1)テストステロンの脂肪肝抑制作用(図4)
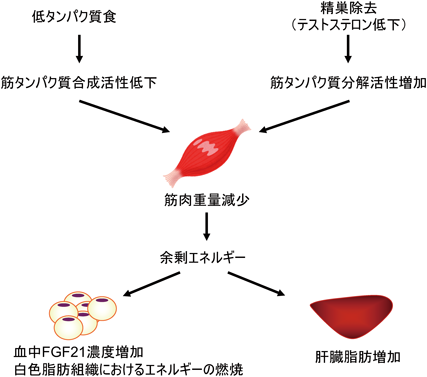
性成熟前(3週齢)と性成熟後(8週齢)の雌雄のラットにタンパク質食を20%含む対照食あるいは5%含む低タンパク質食を7日間与えたところ,タンパク質欠乏による肝臓脂肪増加が,性成熟前のラットでは雌雄同様に生じたのに対し,性成熟後は雌と比較して雄で小さかった.この結果は性成熟に伴って雄で脂肪肝が生じにくくなることを示唆しており,雄性ホルモンによる脂肪肝抑制作用が想定された.
そこで,タンパク質欠乏による脂肪肝に雄の性ホルモンであるテストステロンが及ぼす影響を,精巣除去ラットを用いて解析した.5週齢の雄ラットに偽手術あるいは精巣除去手術を施し,術後回復期間を経て,タンパク質を20%含む対照食あるいは3%含む低タンパク質食を与えて9日間飼育した.その結果,偽手術群では低タンパク質食摂取による肝臓脂肪増加が生じなかったが,精巣除去群では肝臓脂肪が増加した.したがって,性成熟後の雄でタンパク質欠乏による脂肪肝が生じにくいのは,内因性テストステロンが肝臓への脂肪蓄積を抑制するためであることが明らかになった.
テストステロンの主な標的臓器が筋肉であることから,雄では低タンパク質食摂取による筋肉量の低下をテストステロンが抑制することで,筋肉のエネルギー消費を高く維持し,脂肪肝を抑制するのではないかと考えて検討を行った.筋肉タンパク質量は低タンパク質食と精巣除去の条件が重なることで有意に低下した.この要因として,タンパク質欠乏によって生じる血中IGF-I濃度の低下と筋タンパク質合成活性の低下,精巣除去によって生じるオートファジー活性の増加が同時に起こることで筋肉タンパク質量が低下することを明らかにした.肝臓脂肪の増加も低タンパク質食摂取と精巣除去の条件が重なったときに生じることから,テストステロンによる筋肉量の維持が雄の脂肪肝形成を抑制するものと考えられた.「2. ホルモンによる調節」ではIGF-I低下による筋肉量の低下はタンパク質欠乏時の脂肪肝の主要因にならないことが示されたが,テストステロン欠乏による筋肉量の低下はタンパク質欠乏による脂肪肝形成を促進し,脂肪肝形成の性差の要因となることが明らかになった.タンパク質欠乏による脂肪肝発症のリスクに性差があるという結果は,ヒトの健康を考える上でも重要な知見である.さらに,雄でも筋重量の低下が脂肪肝のリスクを高めるという点で,脂肪肝の制御に筋肉が重要な役割を果たすことを示す結果であると考えている
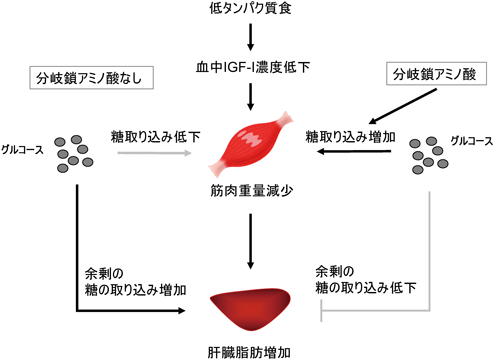
2)分岐鎖アミノ酸の脂肪肝抑制作用(図5)
食餌中のアミノ酸がタンパク質栄養状態の悪化による脂肪肝に及ぼす影響について検討する過程で,低タンパク質食への分岐鎖アミノ酸添加が脂肪肝を抑制するという現象を見いだした.タンパク質含量が3%の低タンパク質食に,3種類の分岐鎖アミノ酸すべてを対照食(タンパク質含量15%)レベルまで添加すると脂肪肝が生じなくなった.同量のグルタミン酸を対照として添加した場合には脂肪肝が抑制されなかったことから,分岐鎖アミノ酸にはタンパク質欠乏による脂肪肝を抑制する特徴的な作用があると予想された.
分岐鎖アミノ酸添加は肝臓における脂肪酸合成,脂肪酸酸化などの脂質代謝酵素の合成には明確な影響を及ぼさなかった.また分岐鎖アミノ酸には筋肉のタンパク質合成活性化作用があることが知られているため38),分岐鎖アミノ酸添加が筋肉重量や筋タンパク質代謝の変化を介して脂肪肝を抑制する可能性も検討したが,これらの指標の変化では脂肪肝抑制作用を説明することができなかった.一方,分岐鎖アミノ酸による筋肉への糖取り込み促進作用も報告されていたことから39),分岐鎖アミノ酸が臓器への糖取り込みの変化を介して脂肪肝を抑制する可能性を検討した.低タンパク質食あるいは分岐鎖アミノ酸を添加した低タンパク質食を摂取したマウスに2-デオキシグルコースを投与して,臓器への糖取り込み活性をin vivoで測定した.その結果,低タンパク質食摂取時に筋肉の糖取り込みが低下するが,分岐鎖アミノ酸を添加すると筋肉への糖取り込みが回復するとともに肝臓への糖取り込みが減少することが明らかになった.したがって,分岐鎖アミノ酸の添加は筋肉への糖取り込みを増加させることにより,肝臓への糖取り込みを低下させ,肝臓での脂質合成の基質量を減らすことで脂肪肝を抑制しているものと考えられた.筋肉と肝臓の臓器間連携による脂肪肝抑制機構として,余剰エネルギーの移動が糖取り込みの形で制御され,肝臓脂肪合成の基質量の変化を介して脂肪肝形成を制御するという機構を明らかにすることができた.
以上述べてきたように,タンパク質栄養状態の悪化によって起こる肝臓脂肪蓄積には,体内の多様な代謝変化が関わっていることがわかってきた.ホルモン濃度の変化によって,臓器間のエネルギー源(糖・アミノ酸・脂肪酸)の動きに変化が起きると同時に,肝臓内での代謝も変動し,肝臓に過剰な脂肪が蓄積するというメカニズムがみえてきた.さらに,全アミノ酸の不足による脂肪肝形成メカニズムとアルギニンだけの不足によるメカニズムが異なること40),アミノ酸自身がホルモン作用を介さずに代謝を調節し,これが脂肪肝形成に寄与すること41)も明らかになってきた.
低栄養は,現代のわが国の高齢者における健康問題となっている.したがって,低栄養による脂肪肝発症機構を解明することは,超高齢化社会における健康寿命延伸のために重要な課題である.本稿で取り上げたタンパク質欠乏による脂肪肝は,筋肉の代謝活性の低下によって増長されるものであり,タンパク質摂取量が不足しがちで,かつ筋肉量や運動量の低下を伴う高齢者の脂肪肝のモデルとなると考えられる.今後,この動物モデルを用いた研究の進展によって,高齢者の脂肪肝のみならず,アジア人に多い非肥満者の生活習慣病の発症機序の解明も可能となると期待している.
最後に,本稿を発表する機会を与えてくださり,共同研究者でもある加藤久典博士(東京大学大学院農学生命化学研究科),共同研究者である高橋伸一郎博士(東京大学大学院農学生命化学研究科),伯野史彦博士(東京大学大学院農学生命化学研究科)に御礼申し上げます.
引用文献References
1) Younes, R. & Bugianesi, E. (2019) NASH in Lean Individuals. Semin. Liver Dis., 39, 86–95.
2) Kumar, R. & Mohan, S. (2017) Non-alcoholic Fatty Liver Disease in Lean Subjects: Characteristics and Implications. J. Clin. Transl. Hepatol., 5, 216–223.
3) Brock, J.F. (1954) Survey of the world situation on kwashiorkor. Ann. N. Y. Acad. Sci., 57, 696–713.
4) Kumar, V., Deo, M.G., & Ramalingaswami, V. (1972) Mechanism of fatty liver in protein deficiency. An experimental study in the rhesus monkey. Gastroenterology, 62, 445–451.
5) Madi, K., Jervis, H.R., Anderson, P.R., & Zimmerman, M.R. (1970) A protein-deficient diet. Effect on the liver, pancreas, stomach, and small intestine of the rat. Arch. Pathol., 89, 38–52.
6) Truswell, A.S., Hansen, J.D., Watson, C.E., & Wannenburg, P. (1969) Relation of serum lipids and lipoproteins to fatty liver in kwashiorkor. Am. J. Clin. Nutr., 22, 568–576.
7) Flores, H., Pak, N., Maccioni, A., & Monckeberg, F. (1970) Lipid transport in kwashiorkor. Br. J. Nutr., 24, 1005–1011.
8) Tse, E.O., Gregoire, F.M., Reusens, B., Remacle, C., Hoet, J.J., Johnson, P.R., & Stern, J.S. (1997) Changes of islet size and islet size distribution resulting from protein-malnutrition in lean (Fa/Fa) and obese (fa/fa) Zucker rats. Obes. Res., 5, 563–571.
9) Toyoshima, Y., Tokita, R., Taguchi, Y., Akiyama-Akanishi, N., Takenaka, A., Kato, H., Chida, K., Hakuno, F., Minami, S., & Takahashi, S. (2014) Tissue-specific effects of protein malnutrition on insulin signaling pathway and lipid accumulation in growing rats. Endocr. J., 61, 499–512.
10) Horiuchi, M., Takeda, T., Takanashi, H., Ozaki-Masuzawa, Y., Taguchi, Y., Toyoshima, Y., Otani, L., Kato, H., Sone-Yonezawa, M., Hakuno, F., et al. (2017) Branched-chain amino acid supplementation restores reduced insulinotropic activity of a low-protein diet through the vagus nerve in rats. Nutr. Metab. (Lond.), 14, 59.
11) Ozaki, Y., Takeda, T., Akanishi, N., Hakuno, F., Toyoshima, Y., Takahashi, S., & Takenaka, A. (2014) Insulin injection restored increased insulin receptor substrate (IRS)-2 protein during short-term protein restriction but did not affect reduced insulin-like growth factor (IGF)-I mRNA or increased triglyceride accumulation in the liver of rats. Biosci. Biotechnol. Biochem., 78, 130–138.
12) Thissen, J.P., Ketelslegers, J.M., & Underwood, L.E. (1994) Nutritional regulation of the insulin-like growth factors. Endocr. Rev., 15, 80–101.
13) Takahashi, S., Kajikawa, M., Umezawa, T., Takahashi, S., Kato, H., Miura, Y., Nam, T.J., Noguchi, T., & Naito, H. (1990) Effect of dietary proteins on the plasma immunoreactive insulin-like growth factor-1/somatomedin C concentration in the rat. Br. J. Nutr., 63, 521–534.
14) Ozaki, Y., Saito, K., Nakazawa, K., Konishi, M., Itoh, N., Hakuno, F., Takahashi, S., Kato, H., & Takenaka, A. (2015) Rapid increase in fibroblast growth factor 21 in protein malnutrition and its impact on growth and lipid metabolism. Br. J. Nutr., 114, 1410–1418.
15) Inagaki, T., Lin, V.Y., Goetz, R., Mohammadi, M., Mangelsdorf, D.J., & Kliewer, S.A. (2008) Inhibition of growth hormone signaling by the fasting-induced hormone FGF21. Cell Metab., 8, 77–83.
16) Kharitonenkov, A., Shiyanova, T.L., Koester, A., Ford, A.M., Micanovic, R., Galbreath, E.J., Sandusky, G.E., Hammond, L.J., Moyers, J.S., Owens, R.A., et al. (2005) FGF-21 as a novel metabolic regulator. J. Clin. Invest., 115, 1627–1635.
17) Laeger, T., Albarado, D.C., Burke, S.J., Trosclair, L., Hedgepeth, J.W., Berthoud, H.R., Gettys, T.W., Collier, J.J., Munzberg, H., & Morrison, C.D. (2016) Metabolic Responses to Dietary Protein Restriction Require an Increase in FGF21 that Is Delayed by the Absence of GCN2. Cell Rep., 16, 707–716.
18) Fisher, F.M., Kleiner, S., Douris, N., Fox, E.C., Mepani, R.J., Verdeguer, F., Wu, J., Kharitonenkov, A., Flier, J.S., Maratos-Flier, E., et al. (2012) FGF21 regulates PGC-1alpha and browning of white adipose tissues in adaptive thermogenesis. Genes Dev., 26, 271–281.
19) Taniguchi, C.M., Emanuelli, B., & Kahn, C.R. (2006) Critical nodes in signalling pathways: insights into insulin action. Nat. Rev. Mol. Cell Biol., 7, 85–96.
20) Brown, M.S. & Goldstein, J.L. (2008) Selective versus total insulin resistance: a pathogenic paradox. Cell Metab., 7, 95–96.
21) Michael, M.D., Kulkarni, R.N., Postic, C., Previs, S.F., Shulman, G.I., Magnuson, M.A., & Kahn, C.R. (2000) Loss of insulin signaling in hepatocytes leads to severe insulin resistance and progressive hepatic dysfunction. Mol. Cell, 6, 87–97.
22) Biddinger, S.B., Hernandez-Ono, A., Rask-Madsen, C., Haas, J.T., Aleman, J.O., Suzuki, R., Scapa, E.F., Agarwal, C., Carey, M.C., Stephanopoulos, G., et al. (2008) Hepatic insulin resistance is sufficient to produce dyslipidemia and susceptibility to atherosclerosis. Cell Metab., 7, 125–134.
23) Shimomura, I., Matsuda, M., Hammer, R.E., Bashmakov, Y., Brown, M.S., & Goldstein, J.L. (2000) Decreased IRS-2 and increased SREBP-1c lead to mixed insulin resistance and sensitivity in livers of lipodystrophic and ob/ob mice. Mol. Cell, 6, 77–86.
24) Leavens, K.F., Easton, R.M., Shulman, G.I., Previs, S.F., & Birnbaum, M.J. (2009) Akt2 is required for hepatic lipid accumulation in models of insulin resistance. Cell Metab., 10, 405–418.
25) Kubota, N., Kubota, T., Kajiwara, E., Iwamura, T., Kumagai, H., Watanabe, T., Inoue, M., Takamoto, I., Sasako, T., Kumagai, K., et al. (2016) Differential hepatic distribution of insulin receptor substrates causes selective insulin resistance in diabetes and obesity. Nat. Commun., 7, 12977.
26) Li, S., Brown, M.S., & Goldstein, J.L. (2010) Bifurcation of insulin signaling pathway in rat liver: mTORC1 required for stimulation of lipogenesis, but not inhibition of gluconeogenesis. Proc. Natl. Acad. Sci. USA, 107, 3441–3446.
27) Yagi, T., Toyoshima, Y., Tokita, R., Taguchi, Y., Okamoto, Y., Takahashi, S.I., Kato, H., & Minami, S. (2019) Low-protein diet enhances adiponectin secretion in rats. Biosci. Biotechnol. Biochem., 83, 1774–1781.
28) Taguchi, Y., Toyoshima, Y., Tokita, R., Kato, H., Takahashi, S.I., & Minami, S. (2017) Triglyceride synthesis in hepatocytes isolated from rats fed a low-protein diet is enhanced independently of upregulation of insulin signaling. Biochem. Biophys. Res. Commun., 490, 800–805.
29) Toyoshima, Y., Tokita, R., Ohne, Y., Hakuno, F., Noguchi, T., Minami, S., Kato, H., & Takahashi, S. (2010) Dietary protein deprivation upregulates insulin signaling and inhibits gluconeogenesis in rat liver. J. Mol. Endocrinol., 45, 329–340.
30) Lin, T.A., Kong, X., Haystead, T.A., Pause, A., Belsham, G., Sonenberg, N., & Lawrence, J.C. Jr. (1994) PHAS-I as a link between mitogen-activated protein kinase and translation initiation. Science, 266, 653–656.
31) Pause, A., Belsham, G.J., Gingras, A.C., Donze, O., Lin, T.A., Lawrence, J.C. Jr., & Sonenberg, N. (1994) Insulin-dependent stimulation of protein synthesis by phosphorylation of a regulator of 5′-cap function. Nature, 371, 762–767.
32) Ma, X.M. & Blenis, J. (2009) Molecular mechanisms of mTOR-mediated translational control. Nat. Rev. Mol. Cell Biol., 10, 307–318.
33) Ricoult, S.J. & Manning, B.D. (2013) The multifaceted role of mTORC1 in the control of lipid metabolism. EMBO Rep., 14, 242–251.
34) Tsukiyama-Kohara, K., Poulin, F., Kohara, M., DeMaria, C.T., Cheng, A., Wu, Z., Gingras, A.C., Katsume, A., Elchebly, M., Spiegelman, B.M., et al. (2001) Adipose tissue reduction in mice lacking the translational inhibitor 4E-BP1. Nat. Med., 7, 1128–1132.
35) Teleman, A.A., Chen, Y.W., & Cohen, S.M. (2005) 4E-BP functions as a metabolic brake used under stress conditions but not during normal growth. Genes Dev., 19, 1844–1848.
36) Toyoshima, Y., Yoshizawa, F., Tokita, R., Taguchi, Y., Takahashi, S.I., Kato, H., & Minami, S. (2020) A translation repressor, 4E-BP1, regulates the triglyceride level in rat liver during protein deprivation. Am. J. Physiol. Endocrinol. Metab., 318, E636–E645.
37) Kilberg, M.S., Shan, J., & Su, N. (2009) ATF4-dependent transcription mediates signaling of amino acid limitation. Trends Endocrinol. Metab., 20, 436–443.
38) Anthony, J.C., Yoshizawa, F., Anthony, T.G., Vary, T.C., Jefferson, L.S., & Kimball, S.R. (2000) Leucine stimulates translation initiation in skeletal muscle of postabsorptive rats via a rapamycin-sensitive pathway. J. Nutr., 130, 2413–2419.
39) Doi, M., Yamaoka, I., Nakayama, M., Mochizuki, S., Sugahara, K., & Yoshizawa, F. (2005) Isoleucine, a blood glucose-lowering amino acid, increases glucose uptake in rat skeletal muscle in the absence of increases in AMP-activated protein kinase activity. J. Nutr., 135, 2103–2108.
40) Otani, L., Nishi, H., Koyama, A., Akasaka, Y., Taguchi, Y., Toyoshima, Y., Yamanaka, D., Hakuno, F., Jia, H., Takahashi, S.I., et al. (2020) Low-arginine and low-protein diets induce hepatic lipid accumulation through different mechanisms in growing rats. Nutr. Metab. (Lond.), 17, 60.
41) Nishi, H., Yamanaka, D., Kamei, H., Goda, Y., Kumano, M., Toyoshima, Y., Takenaka, A., Masuda, M., Nakabayashi, Y., Shioya, R., et al. (2018) Importance of Serum Amino Acid Profile for Induction of Hepatic Steatosis under Protein Malnutrition. Sci. Rep., 8, 5461.