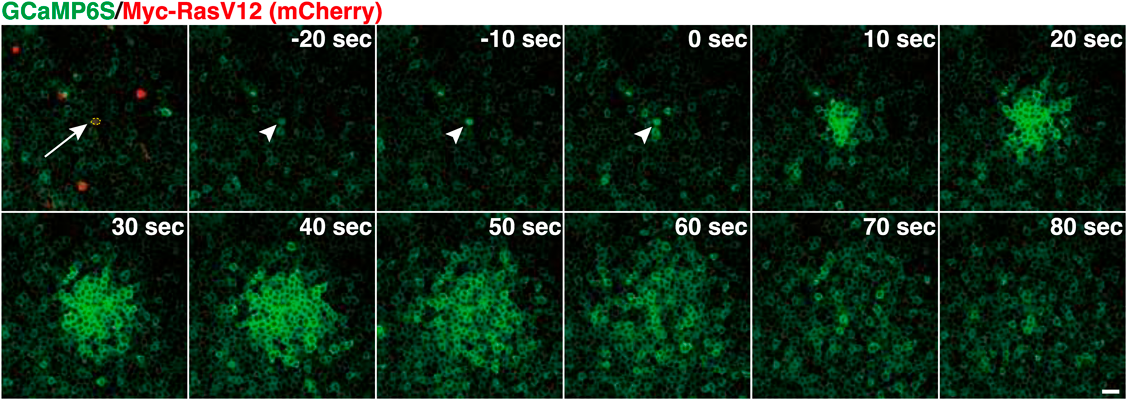
細胞競合における上皮恒常性維持機構の役割Role of epithelial homeostasis in cell competition
1 金沢大学がん進展制御研究所分子病態研究分野Cancer Research Institute of Kanazawa University, Division of Cancer Cell Biology ◇ 〒920–1192 石川県金沢市角間町 ◇ Kakuma-machi, Kanazawa, Ishikawa 920–1192, Japan
2 京都大学大学院医学研究科分子生体統御学講座分子腫瘍学分野Department of Molecular Oncology, Kyoto University Medical School ◇ 〒606–8501 京都市左京区吉田近衛町 ◇ Sakyo-ku, Yoshida-Konoe-cho, Kyoto 606–8501, Japan
発行日:2021年6月25日Published: June 25, 2021