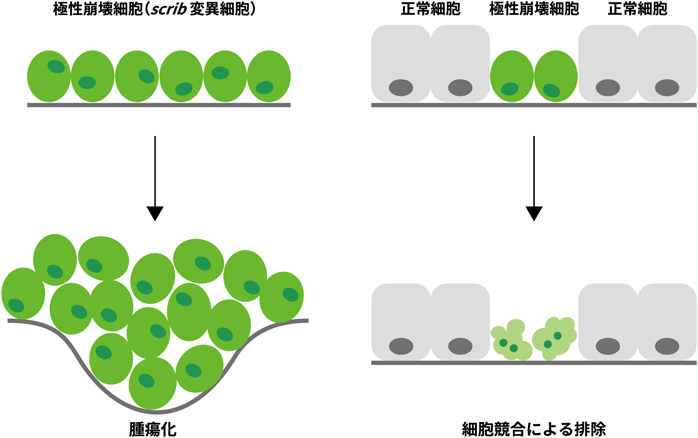
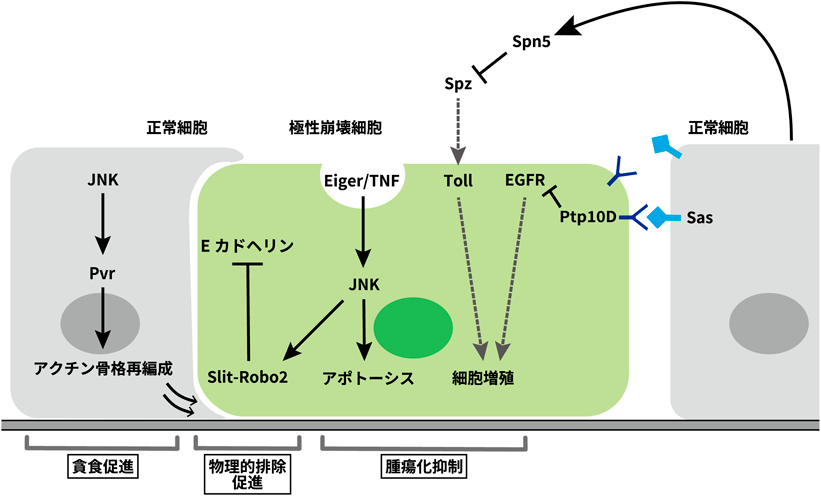
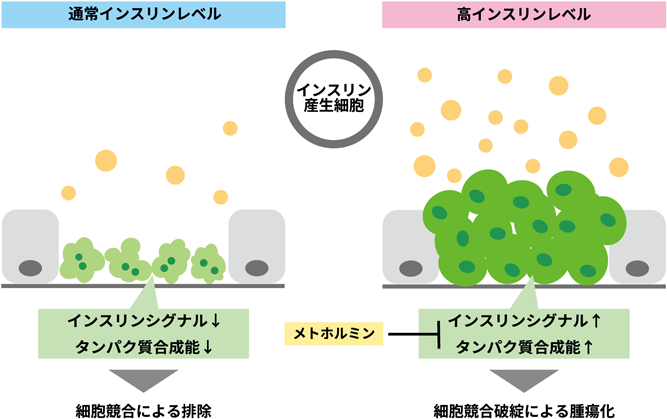
システミックな細胞競合制御によるがん発生メカニズムSystemic regulation of tumor-suppresive cell competition
京都大学大学院生命科学研究科・システム機能学分野Laboratory of Genetics, Graduate School of Biostudies, Kyoto University ◇ 〒606–8501 京都市左京区吉田下阿達町46–29 ◇ 46–29 Yoshida-Shimoadachi-cho, Sakyo-Ku, Kyoto 606–8501, Japan
発行日:2021年6月25日Published: June 25, 2021