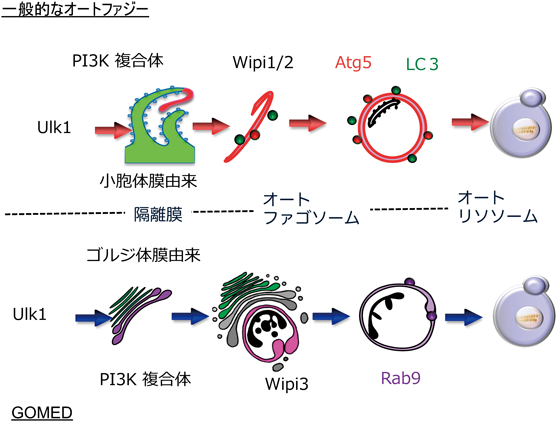
ゴルジ体は,分泌タンパク質や細胞膜タンパク質を目的の場所に適切に運搬するために,糖鎖付加などのタンパク質修飾を行うなど,細胞内の分子輸送を担っている細胞小器官(オルガネラ)である.これらのタンパク質は,小胞体で合成され,ゴルジ体のシス槽,メディアル槽,トランス槽を順に経由する間に,適切な修飾を受け,適切な目的地が設定され,小胞輸送によって運搬される.このようなゴルジ体の古典的な機能に加えて,我々は最近,ゴルジ体膜を利用した新たな細胞内タンパク質分解機構を発見し,Golgi membrane-associated degradation(GOMED)pathwayと命名した.GOMEDは,その形態学的な特徴や機能の類似性から,当初,新規オートファジー(alternative autophagy)と命名したが,解析が進むにつれて,通常のオートファジーとは多くの面で異なっていることが明らかとなった.形態学的にはオートファジーの一種であるものの,別の細胞機能と考えられることからGOMEDの名称とした.GOMEDは生体におけるさまざまな臓器において,生体の恒常性を維持するために決定的な役割を果たしている.本稿では,GOMEDの発見の経緯を含め,最新の知見を紹介する.
マクロオートファジー(以下,オートファジー)は,細胞内のタンパク質などを大量に分解する機構として詳細な解析がなされている.オートファジーは,オメガソームと命名された小胞体膜の一部を起点として形成される.その初期段階では,まず隔離膜と呼ばれる「柿の種」様の構造物が形成される.この隔離膜は伸長するとともに湾曲し,細胞質やオルガネラを囲い込み,最終的には両端が融合して二重膜構造のオートファゴソームができ上がる.オートファゴソームが形成されると,その後にリソソームが直接融合してオートリソソームとなり,囲い込まれた内容物が,リソソームに含まれる消化酵素によって分解される1, 2)(図1).リソソームには,カテプシンなどのタンパク質分解酵素,リソソーム酸性リパーゼ,DNase IIなどさまざまな種類の消化酵素が含まれているため,内容物はほぼ完全に分解される.このようなオートファジーが実行されるためには,Atg5やAtg7などのコア実行分子が必要であると考えられている.
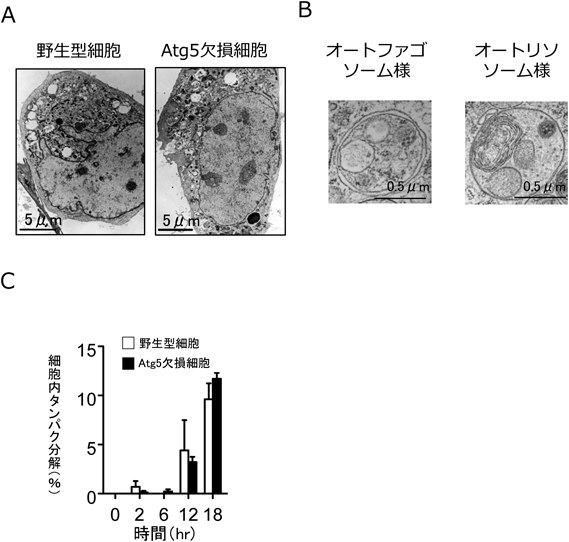
我々は,このようなオートファジーを解析するために,野生型マウスとAtg5欠損マウスより細胞を調製し,まずラパマイシン(mTor阻害を介してオートファジーを誘導する)を投与したところ,野生型細胞においてオートファジー構造やタンパク質分解が認められた.一方,Atg5欠損細胞においてはこのような反応はまったく認められず,Atg5依存的にオートファジーが実行されていることが確認された.ところが,同じ細胞にエトポシド(DNA傷害誘導剤)を投与したところ,Atg5欠損細胞においても,野生型細胞とほぼ同程度に,隔離膜,オートファゴソーム,オートリソソームが出現し,それによるタンパク質分解も認められた(図2).すなわち,Atg5不存在下でも,オートファジー様構造とそれによるタンパク質分解が生じることが見いだされたのである3).de Duveが定義づけたオートファジー4)は,二重膜で囲まれたオートファゴソームの形成と,その内容物のリソソームによる融解であるため,エトポシド投与で出現したAtg5非依存性のオートファジー様構造も,オートファジーの定義に合致するものであった.
Atg5非依存性のオートファジー様構造体について解析を進めたところ,通常のオートファジーとの共通点としては,隔離膜,オートファゴソーム,オートリソソームの細胞内構造の形態がほぼ同一であることが見いだされた(図2).また,酵母細胞から哺乳動物細胞まで進化的に保存されている点も共通していた.一方,相違点に関しては,誘導刺激が異なるほか,オートファジー膜の起源や主な実行分子が異なっていた.特に重要な点は,分解される基質分子が大きく異なっていることであり,このために生体における役割も異なっていた.すなわち,オートファジーの亜型というより,独立した細胞機能であると考えられた.
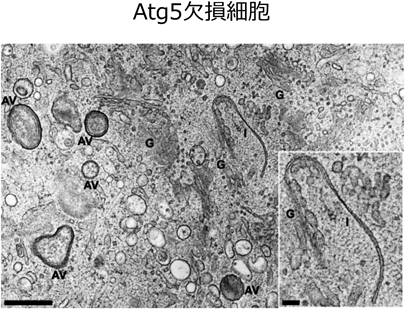
Atg5に依存したオートファジーの膜は小胞体を起源とすることが広く知られている5).しかしながら,Atg5欠損細胞にエトポシドを投与したときに形成されるオートファジーの起源は,ゴルジ体膜に由来する.これは,超微形態学的には,①小胞体の形態に変化がないこと,②ゴルジ体はミニスタック化し,ゴルジ体のトランス側(トランス・ゴルジ)から隔離膜様構造体の形成が認められること,③オートファゴソームの形成において,隔離膜とゴルジ体輸送小胞/エンドソームとの融合が見られること,④オートファジー様構造物はすべてミニスタックゴルジ体に接して存在すること,などの知見3)によって支持されている(図3).さらに,④Brefeldin Aを用いてゴルジ体を分散させると,オートファジー様構造物が認められなくなること,⑤トランス・ゴルジやエンドソームの融合を担っている低分子量Gタンパク質Rab9が,オートファゴソーム/オートリソソーム上に存在していること,⑥Rab9の発現を抑制するとオートファジー様構造がみられなくなること,などの知見が得られ3),Atg5非依存性のオートファジー様構造は,ゴルジ体膜に起源し,Rab9を介した膜融合によって実行されているものと考えられた(図1).
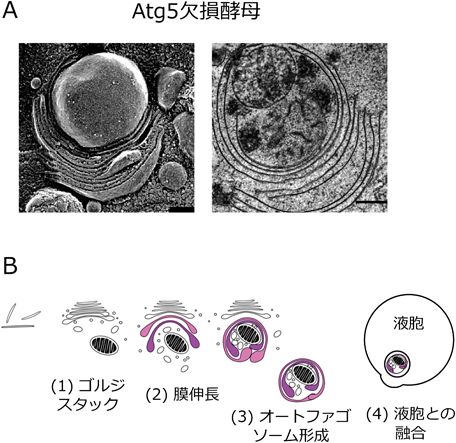
Atg5非依存性のオートファジー様構造は,酵母においても観察される.Atg5欠損酵母細胞に,抗真菌薬のアンホテリシンB1を投与すると,(1)細胞質中のオートファゴソーム形成,(2)オートファゴソームと液胞(哺乳動物細胞のリソソームに相当)との融合が出現する6).このオートファジー様構造も,哺乳動物細胞と同様にゴルジ体膜を起源としている.酵母(Saccharomyces cerevisiae)のゴルジ体は通常細胞質中に散在しているが,アンホテリシンB1を投与すると,ゴルジ体が集積・多層化(スタック形成)し,その遠心部でトランス・ゴルジ膜が湾曲して細胞質成分やミトコンドリアを包み込むオートファゴソームが出現する(図4).実際に,ゴルジ体のスタック形成に関わる分子であるGRH1や湾曲に関わる分子であるGRASP65を欠損した酵母細胞では,オートファゴソーム様構造の形成が起こらない.また,免疫電子顕微鏡において,オートファゴソーム膜上にゴルジ体分子が確認できることより,ゴルジ体膜を起源とするオートファジー様構造であることは明らかである6).これらの形態学的特徴より,Golgi membrane-associated degradation(GOMED)pathwayという名称を付した6).
オートファジーの分子機構は,酵母から哺乳動物細胞までよく保存されており,初期段階においてはセリン-トレオニンキナーゼであるUlk1(酵母ではAtg1)やAtg9, Beclin 1(酵母ではAtg6;PI3キナーゼclass III複合体の構成分子)などの分子が重要となる(図1).続いて起こる隔離膜の伸長には,lipid transfer活性を有するWipi1もしくはWipi2分子が必要であり,さらにその下流で2種類のユビキチン様の結合システムが重要な役割を果たしている.一つは,Atg5とAtg12の共有結合を中心とするAtg5システム(Atg5, 7, 10, 12, 16により機能する)である.Atg5–12複合体は隔離膜の外膜に偏って分布し隔離膜の伸長を促し,オートファゴソームが形成される前後に膜から離脱する.もう一つは,LC3(酵母ではAtg8)とホスファチジルエタノールアミン(PE)の共有結合を中心とするLC3システム(Atg3, 4, 7, 8により機能する)である.LC3-PE複合体は,Atg5–12複合体依存的に隔離膜やオートファゴソーム膜に結合し,オートファゴソーム形成に寄与する1, 2)(図1).
形態学的相同性から,これらの複数の分子がGOMEDにも関与しているものと考えられる.実際に,種々のオートファジー関連分子をノックダウンしたところ,Ulk1やBeclin 1などオートファジー機構の比較的上流で機能する分子群は,GOMEDにおいても重要な役割を果たしていた3).一方,Atg5, Atg7, Atg9, Atg12, Atg16, LC3などのオートファジーコア分子は,GOMEDには関わっていない3).
キナーゼであるUlk1とホモログのUlk2は,オートファジーの初期反応において重要な役割を果たしている.すなわち,Ulk1(あるいはUlk2)のキナーゼ活性を介したAtg13, Fip200との複合体形成が,隔離膜形成に重要である7).一方,Ulk1/Ulk2を欠損した細胞ではオートファジーのみならず,GOMEDも誘導されない3).すなわち,Ulk1(あるいはUlk2)は両方のタンパク質分解機構に関わっていることは明らかである.では,単一の分子が,一つの細胞の中でどのように異なる細胞機能を制御しているのであろうか?
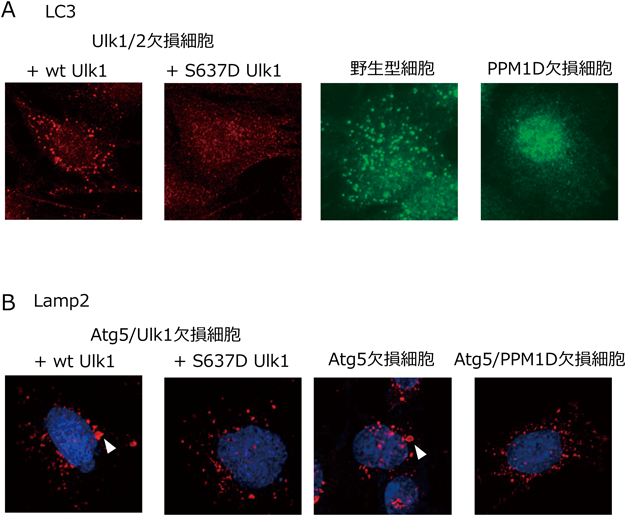
我々は,DNA傷害を与えた細胞を用いて,Ulk1分子の網羅的リン酸化マススペクトルを行うことにより,以下のような事実を見いだすことに成功した8, 9).すなわち,①オートファジー実行時やGOMED実行時には,Ulk1の637番目のセリンが脱リン酸化されること,②セリン残基のアスパラギン酸置換体を作製して,脱リン酸化を妨げると,これらのタンパク質分解系が動かないこと(図5),③この脱リン酸化反応は,DNA傷害後にp53依存的に転写誘導されるホスファターゼPPM1Dが行っていることを見いだした.これらの所見は,PPM1D欠損細胞において,どちらのタンパク質分解系も誘導されないことより確認されている(図5)8).
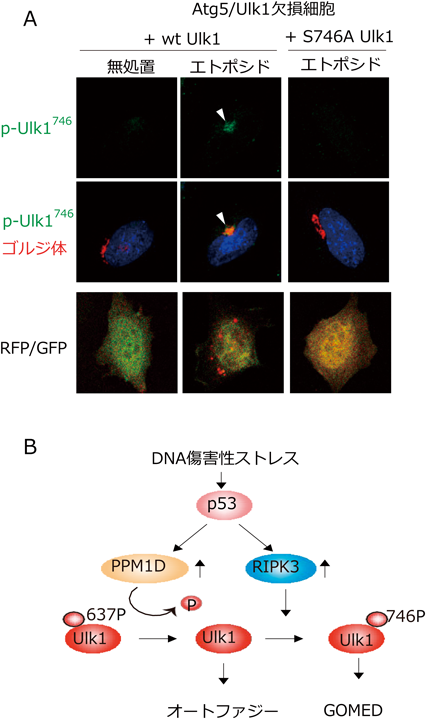
さらに,リン酸化マススペクトルの解析を進めたところ,④GOMED時には,上記の脱リン酸化反応の後に,Ulk1の746番目のセリンがリン酸化されること,⑤セリン残基のアラニン置換体を作製してリン酸化を妨げると,GOMEDのみが動かずオートファジーは誘導されること(図6A),⑥このセリン残基のリン酸化は,p53依存的に転写誘導されるキナーゼRIPK3が行っていることを見いだした9).これらの所見は,DNA傷害刺激を加えたRIPK3欠損細胞においてGOMEDが誘導されないことなどにより明らかであった9).
以上より,DNA傷害によるシグナル伝達機構は以下のように考えられた.まずp53依存的にPPM1DとRIPK3の発現が上昇する.PPM1Dによって,Ulk1の637番目のセリン残基が脱リン酸化されると,オートファジーならびにGOMEDが誘導されうる状態となる.その後さらに746番目のセリン残基がリン酸化されるとGOMEDが活性化され,リン酸化されないと通常のオートファジーが実行されるのである(図6B).なお,746番目のセリンがリン酸化されると,通常のオートファジーで見られるUlk1とAtg13, Fip200との複合体形成は起こらず,Ulk1がゴルジ体に移動した9).おそらく,ゴルジ体上で何らかのタンパク質をリン酸化することにより,下流にGOMEDシグナルを伝達しているものと考えられる.
興味深いことに,GOMEDを制御するRIPK3は,細胞死の一つであるネクロプトーシスの必須分子である10)が,TNF-α+z-VAD(カスパーゼ阻害剤)などのネクロプトーシス誘導刺激を加えてもGOMEDは誘導されない9).また,GOMEDを誘導するDNA傷害刺激によってネクロプトーシスが誘導されることもない.さらに,RIPK3以外のネクロプトーシス分子であるRIPK1やMLKLに関しては,GOMEDとの関与はみられない.これらの事実より,ネクロプトーシスとGOMEDはRIPK3を共同で利用しているものの,システムどうしの相互作用はないものと考えられた.
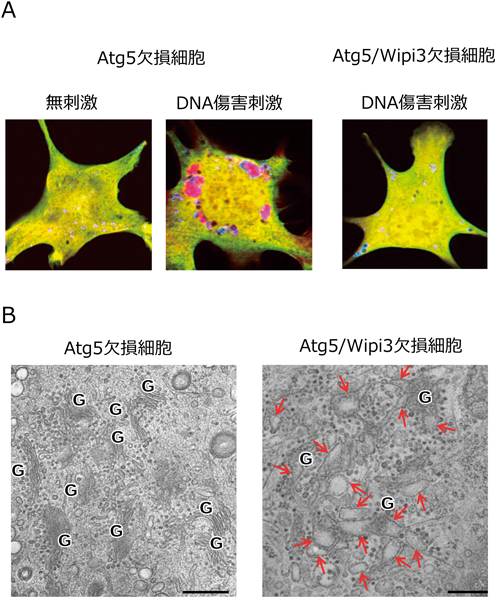
GOMEDは,酵母から哺乳動物まで保存されているため,酵母の遺伝学的手法を用いてGOMEDに必要な遺伝子を探索したところHsv2の同定に至った11).Hsv2はProppinファミリーに含まれるタンパク質で,ホスファチジルイノシトールと結合し,脂質を移動させる機能を有している12).Hsv2を欠損した酵母細胞では,ゴルジ体膜の伸長反応が起こらずGOMEDが誘導されないこと12)より,Hsv2がGOMEDの実行分子であることがわかる.そこで,Hsv2の哺乳動物相同遺伝子であるWipi3を欠損させたAtg5欠損細胞を作製し,DNA傷害刺激を加えたところ,酵母細胞と同様にゴルジ体膜の伸長反応が起こらず,GOMEDは誘導されなかった(図7)12).Wipi3は,通常細胞質に局在しているが,刺激に応じて,またホスファチジルイノシトール3-リン酸(PI3P)依存的にゴルジ体に移動し,ゴルジ体からの隔離膜形成に機能している.Wipi3は,オートファジーの実行に関わるWipi1やWipi2のホモログであるが,ホモログの一方がオートファジーを,他方がGOMEDを制御していることは,両者が異なる機能でありながら,起源を同じくしている可能性がうかがえる.
オートファジーとGOMEDの最大の違いは,分解基質が異なる点にある.オートファジーの基質として有名なp62は,GOMEDで選択的に分解されることはない.逆に,後述するGOMEDが関わるさまざまな生理現象における基質に関しては,オートファジーが関与することはない.すなわち,両者が互いの機能を代償する事例は現時点では認められていない.
GOMEDは,ゴルジ体膜を利用するため,ゴルジ体を経由して運搬される分子が基質分子になることは想像できる.また,この輸送経路に負荷がかかると,ゴルジ体で停滞したタンパク質を分解するために,GOMEDが誘導されることも予想できる.実際に,酵母でGOMEDを誘導するアンホテリシンB1は,エルゴステロールに作用した後に,ゴルジ体から細胞膜へのタンパク質輸送系あるいは細胞外への分泌系を障害することが知られている.アンホテリシンB1以外にも,ゴルジ体から細胞膜,細胞外への輸送に関わる分子を欠損させたときや,細胞外への輸送を阻害する薬剤CBM[1,3-cyclohexanebis(methylamine)]を投与したときに,GOMEDの誘導が観察される6).また,このときに分解される基質分子として,ゴルジ体を経由して運搬される人工タンパク質VSVG-GFP6)や,内在性のインテグリン9)などが見いだされている.これらの知見より,GOMEDの主要な機能は,ゴルジ体から細胞膜/細胞外への輸送が障害されてゴルジ体に滞留したタンパク質を分解することであると考えられた.
このようなGOMED機能の代表的なものとして,インスリン分泌制御がある.インスリンは,膵臓のインスリン分泌細胞(β細胞)で合成されゴルジ体を介して分泌されるが,細胞周囲のグルコース濃度が低下する(すなわち血糖値が下がる)と,さらなる低グルコースを防ぐためにインスリン分泌が抑制される.このときβ細胞内では,GOMEDが誘導されてインスリンの滞留が緩和されている6).この知見は,β細胞株であるMIN6細胞や,マウスから単離した膵島細胞の培養液を高グルコースから低グルコースに変えたときに観察された6).なお,この低血糖時のインスリン分解には,通常のオートファジーは関わっていない.
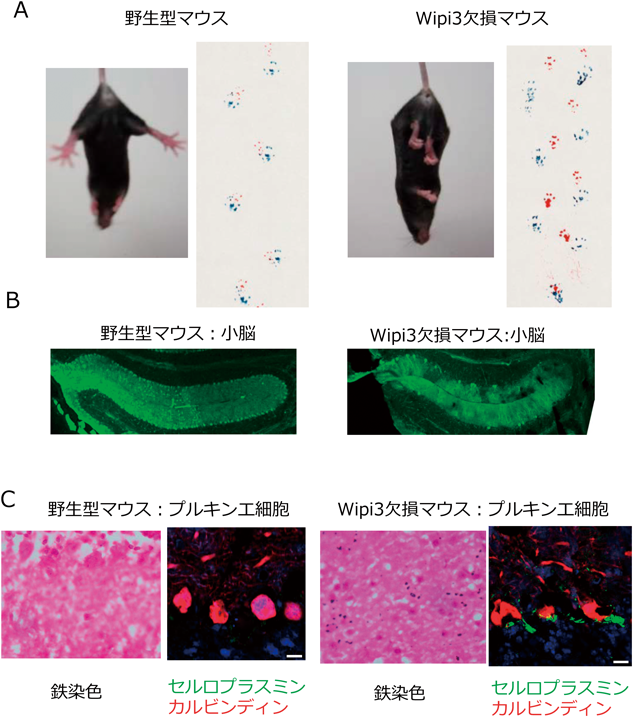
GOMEDの脳での役割を知るために,我々はGOMED実行分子であるWipi3遺伝子を脳特異的に欠損させたマウスを作製した11).このマウスは7週齢ごろより体の姿勢保持が困難となり,歩行障害が生じた(図8A).そこで,運動を制御する小脳を調べたところ,(1)小脳のプルキンエ細胞が変性脱落していること(図8B),(2)脱落前の神経細胞でゴルジ体の形態が異常であること,(3)GOMEDが誘導されない,などの異常が認められた.さらに,詳細な解析を行ったところ,(4)神経細胞に,鉄ならびに鉄輸送タンパク質であるセルロプラスミンが蓄積していること(図8C)が見いだされた11).セルロプラスミンは銅の運搬タンパク質であるが,鉄代謝も行っている.また,肝臓で産生されたセルロプラスミンは血中で機能するが,神経細胞のセルロプラスミンは,ゴルジ体を経由して細胞膜に運搬され,二価鉄を三価鉄に変換する機能を有している13).三価鉄は,フェロポーチンを経由して細胞外に排出される.細胞レベルの検討で,セルロプラスミンがGOMEDの分解基質であることが証明されていることを勘案すると,Wipi3欠損マウスにおいては,GOMEDの異常からその基質分子セルロプラスミンが分解されずに細胞質内に異所性に蓄積し,その結果,三価鉄が過剰になり鉄沈着性の小脳変性疾患を起こしているものと考えられた.
オートファジーに必須のAtg7を欠損したマウスの脳でも同様の神経機能異常が見られる14)が,Wipi3欠損マウスの神経細胞にみられる内部構造とはまったく異なっており,ゴルジ体に異常はなく,小胞体の構造に異常が見られる11).また,セルロプラスミンや鉄の沈着はなく,各々異なる機構で神経細胞の維持に寄与しているものと考えられた.実際に,Wipi3, Atg7の両者を欠損したマウスを作製したところ,生後28日で死亡し,単独の遺伝子欠損マウスよりもはるかに重篤な神経変性を示した11).すなわち,通常のオートファジーとGOMEDは,異なる機構で神経細胞を維持していることが見いだされた.なお,ヒトの脳内鉄沈着神経変性症SENDAは,小児期早期からの非進行性の知的障害と,成人期に急速に進行する錐体外路症状,認知症を呈する神経変性疾患である15)が,Wipi4(Wipi3の相同遺伝子)の遺伝子変異によって発症することが知られている.しかし,Wipi4欠損マウスでは,脳内鉄沈着が見られず,症状もほとんど見られないのに対して,Wipi3欠損マウスでは,脳内鉄沈着が認められ症状も強いため,SENDAのモデルマウスとなりうるかもしれない.
10. GOMEDによる赤血球からのミトコンドリア除去
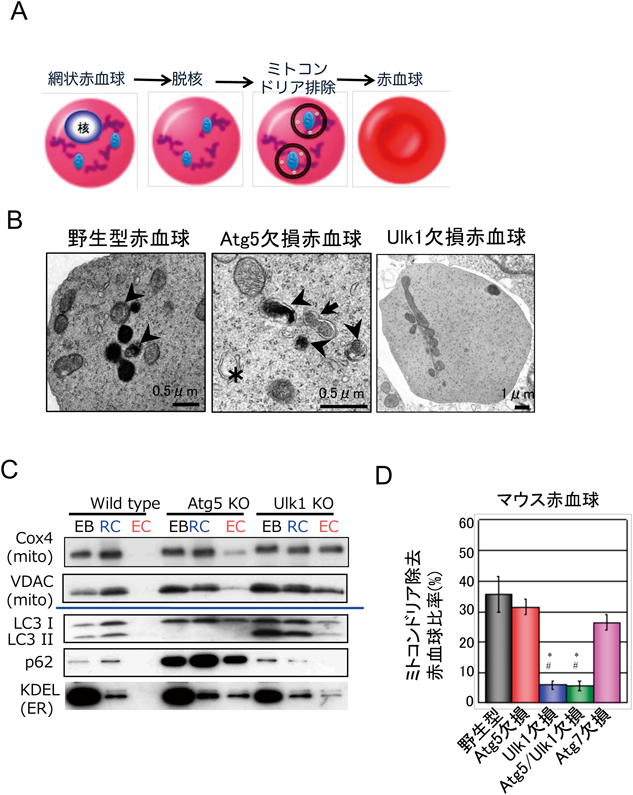
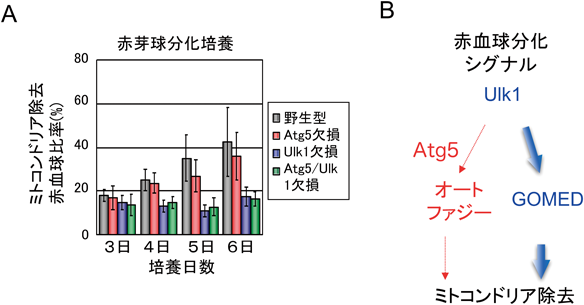
これまで,ゴルジ体を経由する分子がGOMEDの基質となることを紹介してきた.しかし,GOMEDが分解する基質はタンパク質だけでなく,ミトコンドリアなどのオルガネラも分解される.代表的な例が,赤血球分化の最終段階で観察されるミトコンドリアの除去である.赤血球が最終分化する時には,まず脱核が起こり,その後24時間以内にミトコンドリアなどのオルガネラが除去される(図9A).このミトコンドリアの除去機構には,オートファジー様構造体の関与が示唆されていた.実際に,我々が電子顕微鏡を用いて網状赤血球を観察したところ,ミトコンドリアが二重の膜に囲まれているオートファゴソーム様構造,ミトコンドリアの一部の分解が進んでいるオートリソソーム様構造が観察された(図9B).また,この膜の起源は,拡大して観察した形態からゴルジ体膜であると考えられた.Atg5を欠損したマウスにおいても,ミトコンドリアを囲んだオートファゴソーム様構造やオートリソソーム様構造は同程度に観察された(図9B).さらに,ミトコンドリアの多寡を胎仔肝臓(赤血球の造血の場である)と血液で観察したところ,Atg5欠損マウスにおけるミトコンドリア除去は野生型マウスとほぼ同程度に誘導されており,赤血球の多くはミトコンドリアを含んでいなかった(図9C)16).すなわち,Atg5に依存したオートファジーは,赤血球におけるミトコンドリア除去に関わっていないものと考えられた.一方で,オートファジーとGOMEDの両者に関わっているUlk1を欠損したマウスにおいては,赤血球内にミトコンドリアが多数残存していた(図9C).赤血球中の残存ミトコンドリアをより定量的に評価するために,赤血球の分化マーカーTer119, CD71とミトコンドリア(Mitotracker deep red)を三重染色してフローサイトメーターにて測定したところ,正常赤血球やAtg5欠損赤血球,Atg7欠損赤血球ではミトコンドリアが除去されていたが,Ulk1欠損赤血球,Ulk1/Atg5二重欠損赤血球ではミトコンドリアが残存していた(図9D)9).さらに,野生型マウス,Atg5欠損マウス,Ulk1欠損マウス,Atg5/Ulk1二重欠損マウスから,それぞれ赤芽球を調製し,シャーレの中で血球を分化させたところ,野生型赤芽球とAtg5欠損赤芽球では,脱核後にミトコンドリア除去が行われたが,Ulk1欠損赤芽球とAtg5/Ulk1二重欠損赤芽球では,ミトコンドリア除去は行われなかった(図10A)9).これらの解析より,赤血球の最終分化時にみられるミトコンドリア除去は,Atg5依存的なオートファジーではなく,Ulk1に依存したGOMEDに依存していることが証明された(図10B).
本稿では,GOMEDに関して,発見の経緯から,現時点で明らかにされている分子機構,生理機能に関して概説した.ただし,現状において,GOMEDの全体像が明らかになっている訳ではない.具体的には,①GOMED実行メカニズムの詳細,②基質のさらなる同定,③種々の疾患との関連性,など,今後解決していくべき問題は多い.本稿に記したように膵β細胞,赤血球,神経細胞などにおけるGOMEDの役割は明確であるが,GOMEDは,すべての細胞が保有する機能であるために,それぞれの細胞において変調が生じると,さまざまな疾患を発症することが予想される.このような面を明らかにするために,今後は,遺伝子欠損マウスだけではなく,ヒト試料を用いた解析も必要になるものと考えている.
引用文献References
1) Mizushima, N., Levine, B., Cuervo, A.M., & Klionsky, D.J. (2008) Autophagy fights disease through cellular self-digestion. Nature, 451, 1069–1075.
2) Xie, Z. & Klionsky, D.J. (2007) Autophagosome formation: Core machinery and adaptations. Nat. Cell Biol., 9, 1102–1109.
3) Nishida, Y., Arakawa, S., Fujitani, K., Yamaguchi, H., Mizuta, T., Kanaseki, T., Komatsu, M., Otsu, K., Tsujimoto, Y., & Shimizu, S. (2009) Discovery of Atg5/Atg7-independent alternative macroautophagy. Nature, 461, 654–658.
4) Deter, R.L., Baudhuin, P., & de Duve, C. (1967) Participation of lysosomes in cellular autophagy induced in rat liver by glucagon. J. Cell Biol., 35, C11–C16.
5) Tooze, S.A. & Yoshimori, T. (2010) The origin of the autophagosomal membrane. Nat. Cell Biol., 12, 831–835.
6) Yamaguchi, H., Arakawa, S., Kanaseki, T., Miyatsuka, T., Fujitani, Y., Watada, H., Tsujimoto, Y., & Shimizu, S. (2016) Golgi membrane-associated degradation pathway in yeast and mammals. EMBO J., 35, 1991–2007.
7) Wong, P.M., Puente, C., Ganley, I.G., & Jiang, X. (2013) The ULK1 complex: Sensing nutrient signals for autophagy activation. Autophagy, 9, 124–137.
8) Torii, S., Yoshida, T., Arakawa, S., Honda, S., Nakanishi, A., & Shimizu, S. (2016) Identification of PPM1D as an essential Ulk1 phosphatase for genotoxic stress-induced autophagy. EMBO Rep., 17, 1552–1564.
9) Torii, S., Yamaguchi, H., Nakanishi, A., Arakawa, S., Honda, S., Moriwaki, K., Nakano, H., & Shimizu, S. (2020) Identification of a phosphorylation site on Ulk1 required for genotoxic stress-induced alternative autophagy. Nat. Commun., 11, 1754.
10) Zhang, D.W., Shao, J., Kin, J., Zhang, N., Lu, B.J., Lin, S.-C., Dong, M.Q., & Han, J. (2009) RIP3, an energy metabolism regulator that switches TNF-induced cell death from apoptosis to necrosis. Science, 325, 332–336.
11) Yamaguchi, H., Honda, S., Torii, S., Shimizu, K., Katoh, K., Miyake, K., Miyake, N., Fujikake, N., Sakurai, H.-T., Arakawa, S., et al. (2020) Wipi3 is essential for alternative autophagy and its loss causes neurodegeneration. Nat. Commun., 11, 5311.
12) Baskaran, S., Ragusa, M.J., Boura, E., & Hurley, J.H. (2012) Two-site recognition of phosphatidylinositol 3-phosphate by PROPPINs in autophagy. Mol. Cell, 47, 339–348.
13) Hellman, N.E. & Gitlin, J.D. (2002) Ceruloplasmin Metabolism and Function. Annu. Rev. Nutr., 22, 439–458.
14) Komatsu, M., Waguri, S., Chiba, T., Murata, S., Iwata, J., Tanida, I., Ueno, T., Koike, M., Uchiyama, Y., Kominami, E., et al. (2006) Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature, 441, 880–884.
15) Saitsu, H., Nishimura, T., Muramaatsu, K., Kodera, H., Kumada, S., Sugai, K., Kasai-Yoshida, E., Sawaura, N., Nishida, H., Hoshino, A., et al. (2013) De novo mutations in the autophagy gene WDR45 cause static encephalopathy of childhood with neurodegeneration in adulthood. Nat. Genet., 45, 445–449.
16) Honda, S., Arakawa, S., Nishida, Y., Yamaguchi, H., Ishii, E., & Shimizu, S. (2014) Ulk1-mediated Atg5-independent macroautophagy mediates elimination of mitochondria from embryonic reticulocytes. Nat. Commun., 5, 4004.
著者紹介Author Profile
 清水 重臣(しみず しげおみ)
清水 重臣(しみず しげおみ)東京医科歯科大学難治疾患研究所 教授.医学博士.
略歴1958年福井県に生る.84年大阪大学医学部卒業.以後,外科医として臨床に従事.93年に学位取得.94年より大阪大学医学部助手.2000年より大阪大学医学部助教授.06年より現職.
研究テーマと抱負これまでに,細胞死研究とオートファジー研究を行い,アポトーシスがミトコンドリアを介して実行されること,オートファジー細胞死,GOMEDなどを発見してきた.現在は,新たな細胞死の同定やGOMEDの生体応答に注力している.
ウェブサイトhttps://www.tmd.ac.jp/mri/pcb/index.html
趣味クラシック音楽,読書,阪神タイガース.