超硫黄分子は硫黄原子が複数連なった構造を持つ分子であり,同様の構造が有機化合物やタンパク質分子の官能基としても存在する.これらの超硫黄分子および超硫黄官能基は,生体内での存在や生物学的な役割が注目されつつあるが,化学的な観点からも興味深い性質を有しており,超硫黄分子のユニークな生物学的作用や役割は,超硫黄分子・官能基の特徴的な化学反応性に起因する部分が多いと考えられる.超硫黄分子・官能基の化学的特性を理解することは,超硫黄分子・官能基の生物学的な重要性や意味を探る上で基礎となると考えられる.硫黄原子そのものの特性を踏まえながら,生体分子に関連する硫黄化合物と超硫黄分子・官能基の化学的特性や反応性を考察したい.
超硫黄分子は硫黄原子が複数連なって結合した構造を有する分子である.まず,硫黄原子について化学的特性を考察したい.生化学的には,硫黄原子はシステインやメチオニン残基の側鎖チオールやチオエーテル,あるいはコンドロイチン硫酸の硫酸エステルの形などで生体に含まれる元素という印象だろう.基礎化学的観点では,硫黄は16族に属する元素であり,周期表では第三周期に位置する.周期表において同族元素は似た化学的性質を示す傾向があるが,同じ16族元素である酸素と異なり,硫黄は多様な酸化状態と多様な結合価をとり,硫化水素(H2S),硫黄単体(S),チオール(R–SH),スルホン酸(R–SO3H),硫酸(H2SO4)など多様な化合物の形態を持っている.
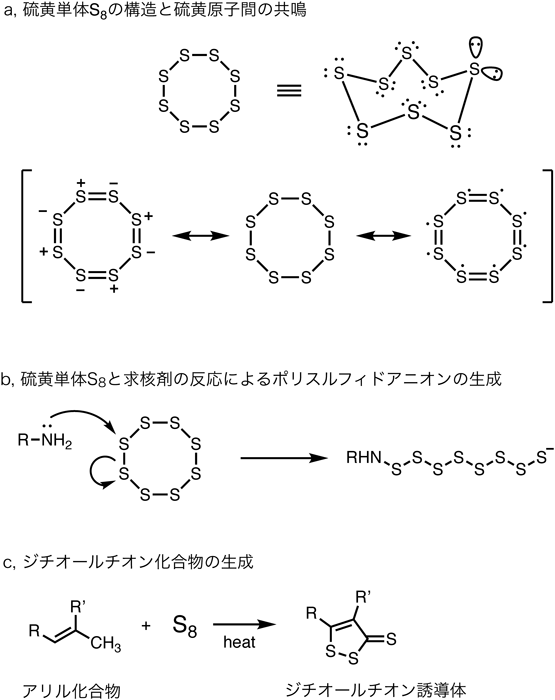
硫黄の単体にはいくつかの形態が存在するが,S8が最も安定な形態として知られており,硫黄原子8個からなる環状のポリスルフィド分子として存在する(図1a).硫黄間の結合(S–S結合)は同族元素である酸素どうしの結合(O–O結合)に比べてかなり安定である.硫黄は第三周期元素であり比較的エネルギーの低い空の3d軌道を有する.この軌道が硫黄原子どうしの結合において,3p軌道電子を受容して共鳴することで安定化に寄与していると考えられる.3d軌道の関与は,酸素に比べて多様な化合物の様態をとりうる要因にもなっており,3d軌道を利用することで,通常の原子価である二価よりも多い原子価(拡張原子価)をとることができる.化学反応の面では,酸素に比べて大きな軌道を有することや,エネルギーレベルが近い3p軌道と3d軌道の相互作用により分極しやすい性質となり,分極誘導を介したいわゆるソフトな反応性を示す.
硫黄原子が連なった構造が超硫黄分子の構造的特徴である点に着目すると,硫黄単体S8は比較的安定なポリスルフィドの一種ということができる.硫黄単体S8の反応性を考察すると,生体内超硫黄分子の反応性を考えるヒントにもなるだろう.
硫黄単体S8は求核剤とも求電子剤とも反応する.アミンや水酸化物イオンなどの求核剤存在下では,S8は求核剤の攻撃を受けて(つまり硫黄原子が求電子的に反応して)開環し,鎖状のポリスルフィドアニオンとなる(図1b).鎖状のS8ポリスルフィドアニオンは非常に不安定で,求核剤の攻撃をさらに受けて反応し硫黄原子数の少ないポリスルフィドアニオンへと分解される.最終的には求核剤が付加したチオラートまで分解される.この点でポリスルフィドは求電子的な化合物であるといえる.
一方,少々ややこしいことであるが,S8ポリスルフィドアニオンや分解中間体のポリスルフィドアニオンは求核性を有している.これにより,系中の他のポリスルフィド分子に求核攻撃することで硫黄原子数が増えたポリスルフィドアニオンを生成する.少量の求核剤存在下のポリスルフィドアニオンは異なる硫黄原子数からなるいくつかの鎖状ポリスルフィドアニオンの平衡混合物の状態ということになる.この点でポリスルフィドは求核的な化合物ともいえる.
電子対を受容して求電子的な反応(求核剤との反応)を起こす一方で,自身の持つ電子対によって求核的な反応(求電子剤との反応)を起こすこともあるという鎖状ポリスルフィドの反応性は非常にユニークであり,またこの性質がポリスルフィドを含む超硫黄分子の反応性の特徴の基礎となっている.前述の硫黄原子間の共鳴(部分的な二重結合性と考えることもできる)により,S8および類縁のポリスルフィドは,硫黄原子一つの化合物(チオール)や酸素類縁体(アルコール)より大きく分極を誘起することができ,電子受容体としても電子供与体としても分極誘導を介するソフトな反応性の求電子剤・求核剤といえる.
硫黄単体S8および類縁のポリスルフィドやポリスルフィドアニオンは,求核剤との反応の他に,ラジカル的な反応も起こすことがある.硫黄ラジカルは同族の酸素ラジカルに比べて安定である.これは前述の硫黄原子の軌道が大きいことが寄与していると考えられる.ポリスルフィドの場合はさらに硫黄原子間の結合が二重結合性を持っていることからラジカル軌道の不対電子を共鳴安定化させる効果が働く.硫黄単体S8はアリール構造を有する化合物と加熱条件下,ラジカル的に反応して安定な芳香族複素環化合物であるジチオールチオン誘導体を生成する(図1c)2).ジチオールチオン構造は,細胞生物学的実験で時々用いられる硫化水素ドナーADT誘導体の部分構造としてとして知られる.
硫黄原子は空の3d軌道に基づく前述のようなユニークな化学的性質を有する.硫黄原子を含む官能基もまた特徴的な性質を示す.生化学分野で代表的な硫黄含有化合物あるいは官能基としては,チオールとスルフィド,ジスルフィドがあげられるだろう.ジスルフィドは最も単純なポリスルフィド構造(パースルフィド構造)と考えることもできる.
チオールはアルコール(R–OH)の類縁化合物として取り上げられることが多い.実際に,アルコールもチオールも水素イオンを遊離して「酸」として作用することが可能であり,また二つの非共有電子対で水素イオン(プロトン)を受容して「塩基」として作用することもでき,類似した性質を持つようにみえる.しかし,チオールの化学的特性はアルコールとはかなり異なっている.チオールの一種であるシステインのスルファニル基(SH基)のpKa値(酸性度)は約8.9であり,一般的なアルコールのpKa値15~17に比べてかなり小さく,酸性度が高くなっている3).これは生理的pH条件での電離度が100万倍ほど異なることになる.この性質により,チオールが中性付近の水溶液中でチオラート(R–S−)型となっている割合はアルコールよりはるかに高い.反応性の観点では中性分子(R–SH)よりもアニオン型分子(R–S−)の方が一般に求核性が高く,チオールは生理的pH条件で一定の割合でチオラート型が生じるためよい求核性を示す.アルコールはこの条件ではほとんど電離していないため高い求核性を発揮するまでには至らない.電離していない状態どうしの比較でもチオールは非電離状態アルコールより求核性が高く(大きな電子軌道による分極誘導の効果が大きいため),チオール・チオラートは生理的条件で非常に有効な求核剤となりうることがわかる.
遊離の硫黄化合物である硫化水素(H2S)は,チオールのアルキル基が水素原子に置き換わった化合物と考えることができ,チオール同様電離する作用がある.硫化水素のpKaは約6.9であり,チオールよりさらに酸性度が高く生理的pHでは半数以上の分子がイオン型(HS−)で存在していることになる4–6).ここで超硫黄分子の一種であるパースルフィド類に着目すると,H2S2では硫化水素より小さい5.0,システインパースルフィド(CysSSH)では4.3となり,pKa値がさらに低下,すなわち酸性度がさらに上昇することがわかる3–6).pKa値から考えると,生理的条件下ではシステインパースルフィドはほとんどがアニオン型(CysSS−)で存在していることになる.酸性度が高いことは,共役塩基(脱プロトン化型)が安定であることを意味しており,共役塩基の求核反応性そのものは低下するのが一般的である(共役塩基が安定して存在できるため).しかし,前述のように分子型とアニオン型ではアニオン型の方が求核性は高い.また,同じpKa値をもつチオール(チオラート)を考えるとき,共鳴構造を有するチオラート(たとえば芳香族チオラート)は,アルキルチオラートより求核性が高いことが知られており,システインパースルフィドアニオンもパースルフィド構造の共鳴が存在するため,アニオン自体の求核性も高まっていると考えられる.システイン残基は同じ条件で数%程度しか電離していないと考えると,パースルフィド型となることでシステインの性質が大きく変化することになるといえるだろう.
硫黄の電気陰性度も有機化学および生化学的な反応性を考える上で着目すべき点である.酸素の電気陰性度は3.44であり,硫黄は2.58である.炭素が2.55であることを考えると,たとえばエーテル(R–O–R′)ではC–O結合はかなり分極して電子の電荷は酸素側に偏っており,炭素の求電子性は高まっているが,C–S結合ではほとんど分極していないと考えられる.一方,イオン化ポテンシャルについて考えると酸素が12.6 eV程度であるのに対し硫黄は10.5 eV程度であり,電子の軌道の分極は起こりやすく,求核剤や求電子剤の接近による分極誘導が起こりやすい.この特性が,求核剤などの反応剤が存在するときに効率よく反応する性質に関わっている.
先ほど,炭素と硫黄の電気陰性度は非常に近い値だと述べたが,そうはいっても硫黄の電気陰性度はわずかであるが炭素より大きくなっているので,硫黄の置換基ではσ効果が働いて弱い電子求引性基として作用する.たとえばメチルチオ基(CH3S基)がパラ位に置換したフェノール誘導体では,フェノールの酸性度がわずかに上昇する.これは,メチルチオ基が電子求引性を発揮してフェノラートのアニオンを安定化するからである.一方,メトキシ基(CH3O基)がパラ位に置換する場合は,フェノール誘導体の酸性度は大きく低下する(pKa値が大きくなる).これは,σ結合を介する効果(σ効果)では酸素は電気陰性度の差から電子求引性基として作用する一方,π結合を介する効果(π効果)では酸素の非共有電子対に由来する2p軌道電子が効率よくベンゼン環のπ電子軌道と共鳴することで強い電子供与性を示すからである.電子を押し出す効果が強ければ,フェノラートのアニオンを安定化できず電離が不利となり,プロトンの解離が起こりにくくなる.硫黄原子の場合も非共有電子対があるので,酸素と同様の効果が生じてもよいと感じるかもしれないが,実際にはそのような効果は小さいことが酸性度の変化からわかる.これは,硫黄の非共有電子対が3p軌道に存在しており,ベンゼン環のπ軌道を形成する2p電子軌道由来の軌道と,軌道の重なりが効率よく起こらず,うまく相互作用できないからである.通常,生体分子も医薬化合物も炭素を主体とした化合物であるので,3p軌道電子で結合を形成する硫黄原子はπ結合性の電子供与の効果は非常に弱く,酸素と違った性質を有することがわかる.このような性質から,チオエーテル化合物(R–S–R)やチオール化合物では硫黄原子が電子の豊富な状態に維持され,酸化されやすい状態となっている.酸化ストレスなど酸化的環境下では,チオールがスルフェン酸(R–SOH)に酸化され,場合によってはさらにスルフィン酸(R–SO2H),スルホン酸へと酸化される.
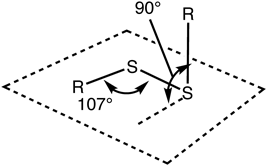
結合角についても,エーテル(C–O–C)とチオエーテル(C–S–C)ではようすが異なる.エーテルのC–O–Cの結合角が117度であるのに対し,チオエーテルのC–S–Cの結合角は105度と見積もられており,硫黄原子の結合性軌道は酸素よりp性が高い(二つのp軌道がなす角である90度に近い)といえる.このことは,ジスルフィドの二面角にも影響している.ジスルフィド結合の二面角はほぼ90度である.置換基の立体障害や非共有電子対の電子反発を想像すると二面角はもっと大きい方が安定のように感じるかもしれない.実際に,同様の構造を有する酸素類縁体であるパーオキシド分子(R–O–O–R′)は大きな二面角を持っている.酸素の場合には,二つの酸素原子の非共有電子対どうしの電子反発や置換基の立体障害が影響するため,パーオキシドに結合する二つの置換基はトランス配置に近い形が安定となる.酸素の場合は非共有電子対も結合価もsp3混成軌道[(2s)1(2p)3]からなっている一方,硫黄の場合は軌道の状況は複雑で,非共有電子対はp軌道,二つの原子価はspd混成軌道から形成されていると考えられる.このため,ジスルフィド結合の二面角(C–S–S–C)はp軌道が互いになす角である90度のときが最も安定となる(図2).また,硫黄単体のS–S結合にもみられるように,非共有電子対の電子の軌道は3d軌道との共鳴が生じるので,酸素の類縁体(パーオキシド)の酸素間結合とは結合のようすも大きく異なり安定化されている.一方,酸化型リポ酸にみられるような小員環の環状のジスルフィドの場合は環状構造による制約のため,上記の最安定な二面角をとることができない場合がある.その場合にはジスルフィド結合の安定性が低下して,反応性が高まることになる.
ジスルフィド結合が拡張された超硫黄構造であるポリスルフィド(R–S–S–S–R′など)については,まだ検討が進んでいないが,硫黄単体S8のポリスルフィドの結合のようすなどから推測すると,ジスルフィドと同様に硫黄原子間の結合周りの二面角はそれぞれ90度に近い角度になっていると考えられる.タンパク質等のジスルフィド結合が拡張された超硫黄構造ではトリスルフィドあるいはテトラスルフィドが比較的安定な構造と推測できるが,上記の硫黄間結合の構造的な特徴を考慮すると,タンパク質上の超硫黄官能基の構造はかなり折りたたまれた形やループ状に曲がった形になっていると想像できる.
ジスルフィド構造は硫黄間結合自体の性質以外にも,特徴的な電子構造に由来して,隣接する構造にも電子的な影響を与える.α位炭素(ジスルフィドの硫黄が結合する炭素原子)上に水素を有するジスルフィド分子(R–CH2–S–S–R′)では,ジスルフィドのd軌道を介する共鳴効果によってα位炭素上の水素の酸性度が上昇する(pKa値が低下する).言い換えるとα位カルボアニオンの安定性が上昇する.トリスルフィド等の超硫黄構造ではこのような効果は確認されていないが,共鳴安定化の効果が増しているので,さらに酸性度が上昇する可能性が考えられる.超硫黄分子の硫黄原子を介する反応ばかりでなく,隣接原子上での反応にも着目する必要があるかもしれない.生化学上重要なジスルフィドあるいはポリスルフィド構造はシスチンおよびシスチン由来の超硫黄構造であるといえるが,これらのポリスルフィド構造からみたα位炭素は,システイン側鎖のメチレン炭素であり,求電子的な反応を起こすことが知られるが,塩基存在下の求核的反応の可能性にも興味が持たれる.
4. ジスルフィド,ポリスルフィドの化学反応性:求核置換反応1)
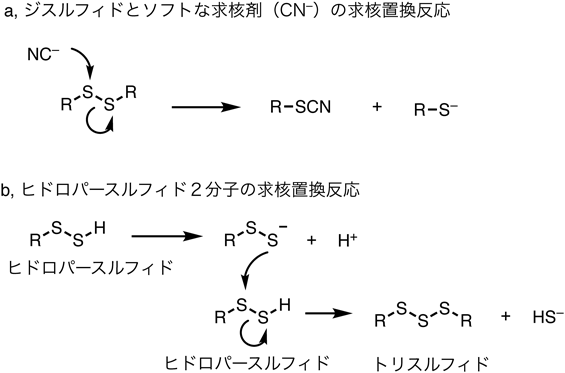
硫黄(スルフィドやチオール)およびポリスルフィドの反応として生化学的に重要なものの一つのは置換反応であろう.ジスルフィドでは,求核剤から(二つの硫黄原子のうちの)一方の硫黄原子に攻撃を受け,S–S結合をイオン的に開裂してチオラート(R–S−)を生じることで求核置換反応を起こす(図3a).たとえば,シアン化物イオンの求核攻撃ではチオシアニド(R–SCN)と対応するチオラート(R–S−)を生じる.求核剤としては,アミンや水酸化物イオンなども反応しうる.ここで,ジスルフィド結合の一方の硫黄原子は求電子的な反応性を示し(求核剤の非共有電子対を受容するから),もう一方の硫黄原子はチオラートとして脱離基の役割を果たしている.
ジスルフィドの一方のアルキル置換基が水素で置き換わったヒドロパースルフィド(R–SSH)も種々の求核剤と置換反応を起こして,R–S–Nu(Nuは求核剤が付加した構造)と硫化水素イオン(HS−)を生じるか,あるいはチオラート(RS−)とNu–SHを生じる.ヒドロパースルフィドはそれ自身も求核性を有すると考えられ,前述のヒドロパースルフィドのpKa値を参照すると,ヒドロパースルフィドは容易に電離してパースルフィドアニオンを生じ優れた求核剤として作用しうると想像できる.この場合には,ヒドロパースルフィドのジスルフィド結合に,別の分子のパースルフィドアニオンが反応して,トリスルフィド(R–SSS–R)を生じることになる(図3b).この反応では2分子のヒドロパースルフィドが関与するが,一方のヒドロパースルフィドは求核剤として反応し,もう一方は求電子剤(求核剤からの電子対を受容する役割)として反応する.このような反応性には,パースルフィドの硫黄原子上の電子が分極しやすく多様な状態をとりうる性質が表れている.
ジスルフィドの求核置換反応における脱離基の性能は,脱離するチオラートの安定性,すなわちチオールの酸性度に対応するので,ジスルフィドに対して求核剤が反応する(すなわちチオールが脱離する)よりも,トリスルフィドあるいはテトラスルフィドに求核剤が反応する方がはるかに早く進行する.なぜなら脱離するパースルフィドアニオン,トリスルフィドアニオンの安定性が非常に高い(すなわち,これらに対応するプロトン化体,つまり共役酸の酸性度が非常に高い)からである.硫黄単体S8も求核剤との反応性が高いことはすでに述べたが,効率よく反応するのは同じ理由である.この点で,ポリスルフィド構造は反応性に富んだ官能基であるといえる.
ジスルフィドの求核置換反応は酸触媒存在下で加速される.これは,(ルイス塩基性は酸素より弱いが)ジスルフィドの一方の硫黄原子の非共有電子対にルイス酸が結合することで3配位のスルホニウム構造が生じ,これにより脱離能が著しく上昇したためと考えられる.一方,ルイス酸が存在しない場合は置換反応は遅くなる.生体内の緩衝液中でもジスルフィド結合の求核置換反応は反応環境のルイス酸の補助により加速されると予想できる.
ジスルフィド結合への求核剤の求核置換反応は,sp3炭素上への求核置換反応と同様に背面攻撃で起こると考えられるが,反応の中間構造においては違いがみられる.典型的なsp3炭素上での求核置換反応では,脱離基と炭素の結合(σ結合)の反結合性軌道に,求核剤が電子対を供与することで5配位様の遷移状態が形成される.一方,拡張原子価をとることができる硫黄上での求核置換反応は,炭素よりも求核剤・脱離基(チオラート)との結合長が長く(S–S結合長は約2 Å),3d軌道が存在することにより,5配位構造の中間体をいったん生成して反応が進行すると考えられる.
ジスルフィドの求核置換反応においては,脱離基(チオラート)の脱離能が反応の速さに影響することはすでに述べた.炭素上の求核置換反応と同様に脱離基の共役酸の酸性度が高い(pKaが小さい)方が有利であるので,脱離するチオラートの硫黄原子上の電子密度が小さくなるような効果,すなわち電子求引性効果を示す官能基がある化合物の方が反応性がよく,逆に電子を供与する官能基では遅くなる.双性イオンの状態のシスチンはσ効果によって,カルボキシラート基からは電子供与の効果を,アンモニウム基からは電子求引の効果を受ける.シスチンのカルボキシ基,アミノ基は環境のpHによって電離状態が変わるので,pHによってジスルフィド結合の求核置換反応の反応性が変化する.塩基性環境下のシスチンのジアニオン型では,カルボキシラートの効果により中性条件下の双性イオン型に比べて200分の1程度まで反応性が低下する(シアン化物イオンとの反応の場合).
5. ジスルフィド,ポリスルフィドの化学反応性:酸化反応1)
典型的なチオール(R–SH)の酸化反応としてジスルフィドへの酸化があげられるが,ヒドロパースルフィド(R–SSH)も同様の酸化反応を起こしてテトラスルフィドを生成する.有機化学的には酸化剤としてヨウ素(I2)を用いることでヒドロパースルフィドからテトラスルフィドを生成するが,アリールヒドロパースルフィドの酸化の場合には,上記のテトラスルフィドの形成のあと1原子脱硫しアリールトリスルフィドが生成する場合がある.
ジスルフィドは,オゾン,N2O4,過酸などの酸化剤と反応してスルホン酸無水物やチオールスルホナート(R–S–SO2–R′)が生じる.これらの酸化剤は親電子剤として(硫黄原子の非共有電子対の電子が酸化剤に供与される形で)反応すると考えられる.この反応ではジスルフィドは求核的な性質が表れている.
ヒドロパースルフィド(R–SSH)やヒドロポリスルフィド(R–SnSH:nは自然数)は,化学的には熱分解によりホモリシスを起こしてチイルラジカル(R–S·)とHS·に開裂する.結合解離エネルギーは,パーオキシド(R–O–O–R)が40 kcal/mol程度なのに対し,ジスルフィド(R–S–S–R)が70 kcal/mol程度であり結合エネルギーとしてはパーオキシドより開裂しにくいが,生成するチイルラジカルは対応する酸素同族体であるオキシラジカル(R–O·)と比較して安定である.ポリスルフィドラジカル(R–SnS·)はチイルラジカルと比較してもさらに安定である.ポリスルフィドラジカルは硫黄単体(S8)の熱分解などで生じるが,たとえばオレフィンのラジカル重合反応にポリスルフィドラジカルが共存すると,ラジカル反応を促進するのではなく抑制する方向に作用する.これはポリスルフィドラジカルが非常に安定であって,非ラジカル単量体からのラジカル生成を行うよりも,ポリスルフィドラジカルとオレフィン由来ラジカルの終結反応の方が起こりやすいからだと考えられる.
ジスルフィド結合に少量のスーパーオキシドが反応すると,ラジカル的に反応が進行し,一つの硫黄原子にスーパーオキシドアニオンが付加した生成物(R–S–OO−)とチイルラジカル(R–S·)が生成すると考えられる.チイルラジカルは効率よくジスルフィド結合のラジカル開裂を誘起できるため(k=2×106 M−1 s−1),連鎖的にジスルフィド結合のラジカル開裂と再結合を繰り返す.生化学的観点からは,スーパーオキシドが発生することでタンパク質ジスルフィド結合の組換えが起こる可能性があるといえる.
一方,スーパーオキシドは多量に存在するとジスルフィドに対する求核剤としての反応も起こすと考えられる.ジスルフィドに対するスーパーオキシドの求核的な反応からは,ジスルフィドが酸化されたスルフィナート(R–SO2−)やスルホナート(R–SO3−)が生成する.
ジスルフィドの一つの硫黄が酸化された化合物は,チオールスルフィナート(R–SO–S–R)あるいはチオスルホナート(R–SO2–S–R)と呼ばれる化合物になる.ジスルフィド化合物の部分的な酸化によって生じると考えられ,生化学的にも過酸化水素や過酸,一重項酸素で起こると考えられる.チオールスルフィナートやチオスルホナートでは酸化された硫黄原子の電子密度が低下しており,二つの硫黄原子の反応性に大きな差が生じる.チオールスルフィナートに対して,水酸化物イオンのような反応性の高い求核剤を反応させると,酸素化された方の硫黄原子(–SO–のS)に求核攻撃し,R–SO2−とチオラートを生じると考えられる.一方,シアン化物イオンや他の硫黄求核剤(たとえばR–SH)など分極誘導によって求核性を発揮する求核剤では,酸素化されていない方の硫黄原子(–S–)に求核攻撃が起こり,スルフィナートと対応する求核置換生成物が得られる.これは,チオールスルフィナートの二つの硫黄原子のうち,酸化された硫黄原子(–SO–)は電気陰性度の違いから硫黄原子が電子不足となっており,水酸化物イオンのような高い反応性の求核剤の反応を受けやすく,チオールスルフィナートのもう一方の硫黄原子は,分極誘導による求核剤の付加を受けやすい(いわゆるソフトな求核剤とソフトな求電子剤の反応が起こりやすい).また,チオールスルフィナート自体は不均化反応を起こし,ジスルフィドとチオスルホナートが生じる.
チオールスルホナートは求核剤に対する反応性はジスルフィドよりも高まっており,中性条件下でも加水分解(水による求核攻撃)を受ける.たとえばシスチンモノオキシドは中性条件下加水分解され,このときはジスルフィドとスルフェン酸(R–SO2H)が生じ,不均化反応が起こっていることがわかる7).チオール自体もチオールスルホナートに対する求核剤となりうる.チオールの場合は,チオールスルホナートのどちらの硫黄原子にも攻撃しうると考えられ,最終的にはジスルフィドと水を生成する.これはスルフィン酸を中間生成物として生じた後,チオールがスルフィン酸にも攻撃してジスルフィドを形成するからと考えられる.
チオールスルフィナートに対する酸化反応は複雑である.スーパーオキシドによる求核的な酸化反応では,酸素化された方の硫黄原子(–SO–のS)が求核攻撃を受け,スルフェン酸(R–SO2−)とスルホン酸とジスルフィドを生成する.親電子性酸化剤による酸化反応では,より電子密度の高い硫黄(–S–)を酸化し,ジスルホキシド化合物を生じる.
生体内でのポリスルフィドの化学的挙動はほとんど検討されていない8).ポリスルフィドの化学的特性を考えると,生体内で想定される求核剤,求電子剤,ラジカル化合物との反応性を検討することで,生体内での挙動を推定するのが現時点では妥当であろう.
ジスルフィドと求核剤,求電子剤との化学的反応性は前述したとおりだが,その反応性は周囲の環境によって大きく変化することが想定される.ジスルフィド結合の一方の硫黄原子への求核置換反応は,sp3炭素への求核置換反応と同様に求核剤の求核性と脱離するチオラートの脱離能に依存する.塩基性条件下での水酸化物イオンの攻撃によるアルカリ加水分解は,チオラートとスルフェン酸を生成することから,硫黄酸化物の生体内挙動を考える上で興味深い.一方,S–S結合は共鳴安定化されていることを考えると,分極誘導によるソフトな求核剤との反応性に注目すべきと思われる.チオールあるいはチオラートによるジスルフィドへの求核置換反応は,ジスルフィドの異性化反応でありタンパク質の構造変換など生化学的に興味深い性質に関連する.チオールよりもチオラートの方が求核性が高いことから,ジスルフィド結合周囲の環境が塩基性(あるいはチオラート生成を促進するアミノ酸残基の存在)であることで,ジスルフィド結合異性化の速度は速まる.一方,ジスルフィドの硫黄原子はルイス塩基性も有しており,ルイス酸となる(あるいはプロトン供与できる)アミノ酸残基の影響により,脱離能が上昇して求核置換反応が加速することが考えられる.このような環境では,チオールとも高い反応性を示したり,水酸化物イオンとの反応性が向上したりして,ジスルフィド結合の反応性は高まると予想される.
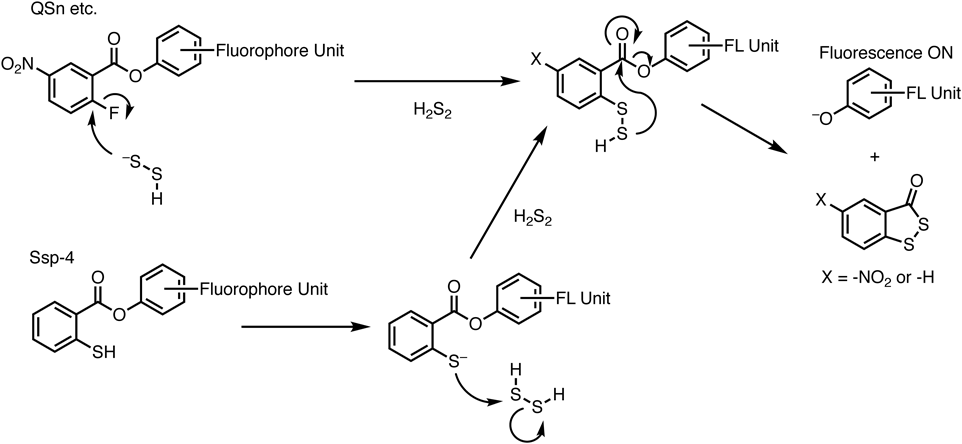
遊離のヒドロポリスルフィド(H2Sn)に対する蛍光プローブがいくつか報告されている(Ssp-4, QSnなど)9–11).これらのプローブ分子では,チオフェノラート(アリールチオラート)がヒドロポリスルフィドに求核置換反応を起こすことで選択的にポリスルフィドを捕捉する仕組み(Ssp-4)や,ポリスルフィドアニオンの求核性により選択的に芳香族求核置換反応を起こす仕組み(QSn)を利用している(図4).チオラートとポリスルフィドの分極を介したソフトな求核剤・求電子剤の反応が生化学的環境下でも効率よく起こることがわかる.
グルタチオンヒドロトリスルフィド(GSSSH)などが生体内で生成することが報告されるなど9),チオラートのポリスルフィド体であるアルキルポリスルフィドアニオンが求核剤として作用する場面も想定される.ポリスルフィドアニオンでも硫黄間結合の共鳴の寄与が増加し,求核反応・求電子反応における分極誘導の効果がより強く表れることから,チオラートよりも複雑な反応性を示すと予想される.前述のように,ヒドロポリスルフィドのpKaは硫黄間結合の共鳴の効果により大きく低下しており,生理的pH条件下ではチオールに比べてポリスルフィドアニオンが生じやすい.求電子剤との反応性や金属配位子としての電子供与性は増大すると考えられる.3-メルカプトピルビン酸硫黄転移酵素(3-MST)という酵素の活性中心におけるシステイン残基の金属配位において,構造解析からシステインパースルフィドアニオン(CysSS−)の配位構造が見いだされており12),チオラート配位よりもより強い電子効果が予想される.
求電子剤としてのポリスルフィドも複雑な反応を起こすと予想される.グルタチオンヒドロパースルフィド(GSSH)やグルタチオンヒドロトリスルフィドは,グルタチオンの求核攻撃を受けることで求核置換反応を起こし,遊離の硫化水素イオン(HS−)やポリスルフィドアニオン(HSnS−),グルタチオンポリスルフィド(GSnG)といった多様な生成物を与えると考えられる.グルタチオンポリスルフィドは,ポリスルフィドとして分極誘導を伴う求核性および求電子性を発揮するため,さらに複雑なポリスルフィド分子を生じうる.
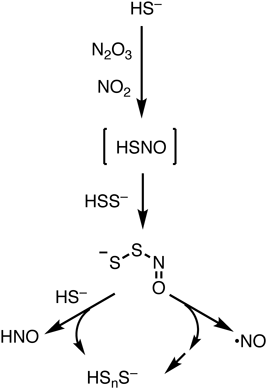
ラジカル分子との反応に着目すると,生体内ではスーパーオキシドや一酸化窒素(ともにラジカル分子)との関連に興味が持たれる.遊離のスルフィドである硫化水素イオン(HS−)は,一酸化窒素(NO)由来のNO2およびN2O3と反応することでHSNOという特徴的な低分子化合物を生成することが報告されている13).HSNOは前述のポリスルフィドアニオン(たとえばHSS−)と反応することで,比較的安定なニトロソパースルフィドアニオン(−SSNO)を生成する.ニトロソパースルフィドアニオンは,一酸化窒素とパースルフィドアニオンに再分解されることが観測されており,一酸化窒素のリザーバーとしての機能があると推定される.また副生するパースルフィドアニオンは,ポリスルフィド特有の求核性を有すると考えられ,自己反応や他のスルフィド分子と反応を繰り返してポリスルフィドを再生すると考えられる(図5)13).これらの反応は,生体内環境では小さなシグナル分子である一酸化窒素やスルフィドが密接に相互作用していることを想起させる.
ポリスルフィドをはじめとした超硫黄分子・官能基の反応性は,生理的条件化での試験管内反応研究や有用なプローブの開発に基づく細胞研究によって今後解明されると思われるが,硫黄の特徴的な化学的特性を踏まえて研究を進めることが重要と思われる.
謝辞Acknowledgments
本稿で参照した硫黄の基本的化学特性は,先達の古典的硫黄化学研究の成果を参照したものである.特に大饗茂先生の有機硫黄化学の知見・解説に寄るところが大きく,大饗先生による『有機硫黄化学』(化学同人)に基づくものであり,深く感謝の意を表したい.
引用文献References
1) 大饗茂(1982)硫黄有機化学,化学同人.
2) Bottcher, B. & Bauer, F. (1951) Über Trithione, V. Mitteil. Notiz über einige neue Trithione). Ber. Eur. J. Inorg. Chem., 84, 458–463.
3) Cuevasanta, E., Lange, M., Bonanata, J., Coitiño, E.L., Ferrer-Sueta, G., Filipovic, M.R., & Alvarez, B. (2015) Reaction of hydrogen sulfide with disulfide and sulfenic acid to form the strongly nucleophilic persulfide. J. Biol. Chem., 290, 26866–26880.
4) Chen, W., Rosser, E.W., Zhang, D., Shi, W., Li, Y., Dong, W.-J., Ma, H., Hu, R., & Xian, M. (2015) A specific nucleophilic ring-opening reaction of aziridines as a unique platform for the construction of hydrogen polysulfides sensors. Org. Lett., 17, 2776–2779.
5) Hoffmann, M.-R. (1977) Kinetics and mechanism of oxidation of hydrogen sulfide by hydrogen peroxide in acidic solution. Environ. Sci. Technol., 11, 61–66.
6) Schwarzenbach, G. & Fischer, A. (1960) Die acidität der sulfane und die zusammensetzung wässeriger polysulfidlösungen. Helv. Chim. Acta, 43, 1365–1390.
7) Savige, W.E. & Maclaren, J.A. (1966) in Organic Sulfur Compounds (Kharasch, N. ed.), Vol. 2, Chapter 15, Pergamon Press, Oxford.
8) Ida, T., Tomohiro Sawa, T., Ihara, H., Tsuchiya, Y., Watanabe, Y., Kumagai, Y., Suematsu, M., Motohashi, H., Fujii, S., Matsunaga, T., et al. (2014) Reactive cysteine persulfides and S-polythiolation regulate oxidative stress and redox signaling. Proc. Natl. Acad. Sci. USA, 111, 217606–217611.
9) Liu, C., Chen, W., Shi, W., Peng, B., Zhao, Y., Ma, H., & Xian, M. (2014) Rational design and bioimaging applications of highly selective fluorescence probes for hydrogen polysulfides. J. Am. Chem. Soc., 21, 7257–7260.
10) Zeng, L., Chen, S., Xia, T., Hu, W., Li, C., & Liu, Z. (2015) Two-photon fluorescent probe for detection of exogenous and endogenous hydrogen persulfide and polysulfide in living organisms. Anal. Chem., 87, 3004–3010.
11) Chen, W., Liu, C., Peng, B., Zhao, Y., Pacheco, A., & Xian, M. (2013) New fluorescent probes for sulfane sulfurs and the application in bioimaging. Chem. Sci. (Camb.), 4, 2892–2896.
12) Hanaoka, K., Sasakura, K., Suwanai, Y., Toma-Fukai, S., Shimamoto, K., Takano, Y., Shibuya, N., Terai, T., Komatsu, T., Ueno, T., et al. (2017) Discovery and mechanistic characterization of selective inhibitors of H2S-producing enzyme: 3-Mercaptopyruvate sulfurtransferase (3MST) targeting active-site cysteine persulfide. Sci. Rep., 7, 40227.
13) Cortese-Krott, M.M., Kuhnle, G.G.C., Dyson, A., Fernandez, B.O., Grman, M., DuMond, J.F., Barrow, M.P., McLeod, G., Nakagawa, H., Ondrias, K., et al. (2015) Key bioactive reaction products of the NO/H2S interaction are S/N-hybrid species, polysulfides, and nitroxyl. Proc. Natl. Acad. Sci. USA, 112, E4651–E4660.
著者紹介Author Profile
 中川 秀彦(なかがわ ひでひこ)
中川 秀彦(なかがわ ひでひこ)名古屋市立大学大学院薬学研究科教授.博士(薬学).
略歴東京大学薬学部卒業.同大学院博士後期課程修了(1995年).放射線医学総合研究所研究員を経て2004年名古屋市立大学大学院薬学研究科助教授/准教授.13年より現職.10~14年さきがけ研究員併任.
研究テーマと抱負創薬化学とケミカルバイオロジー.一酸化窒素とその関連シグナル分子のケミカルツールを開発し生物応用に結びつけたい.また酵素活性計測蛍光プローブを開発しイメージングや阻害剤スクリーニングを推進したい.
ウェブサイトhttp://www.nagoya-cu.ac.jp/phar/grad/soyaku/iyaku/yakka/