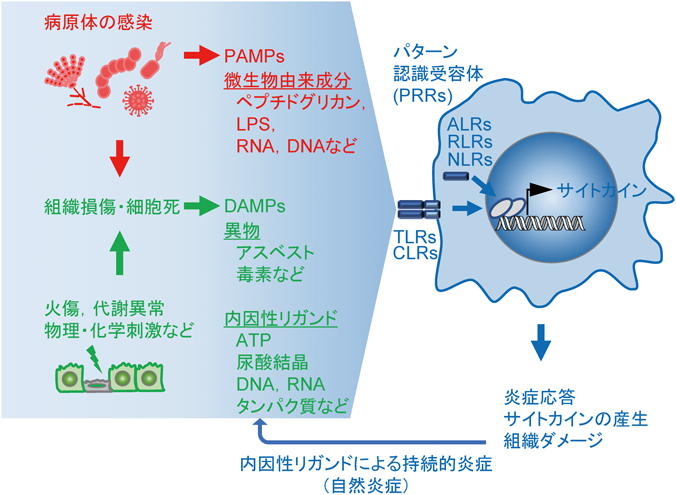
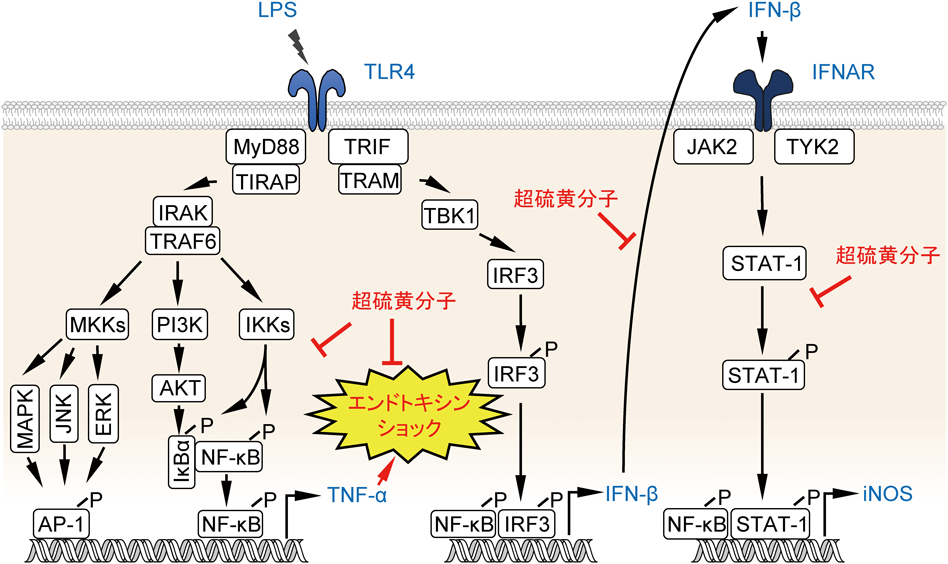
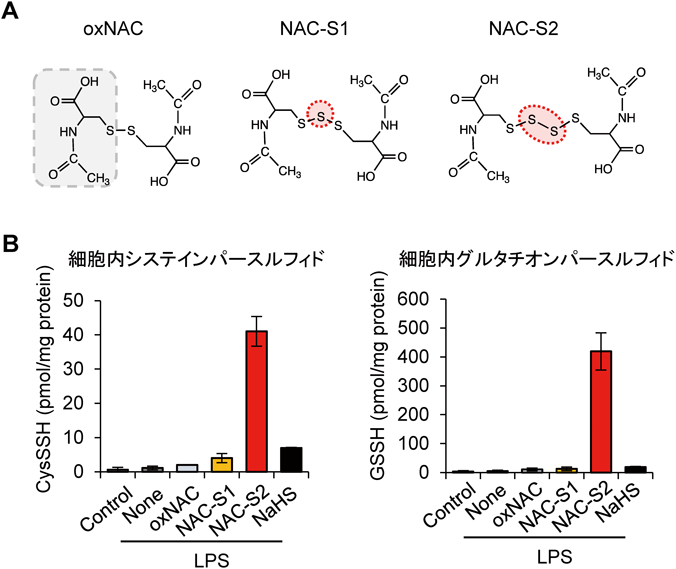
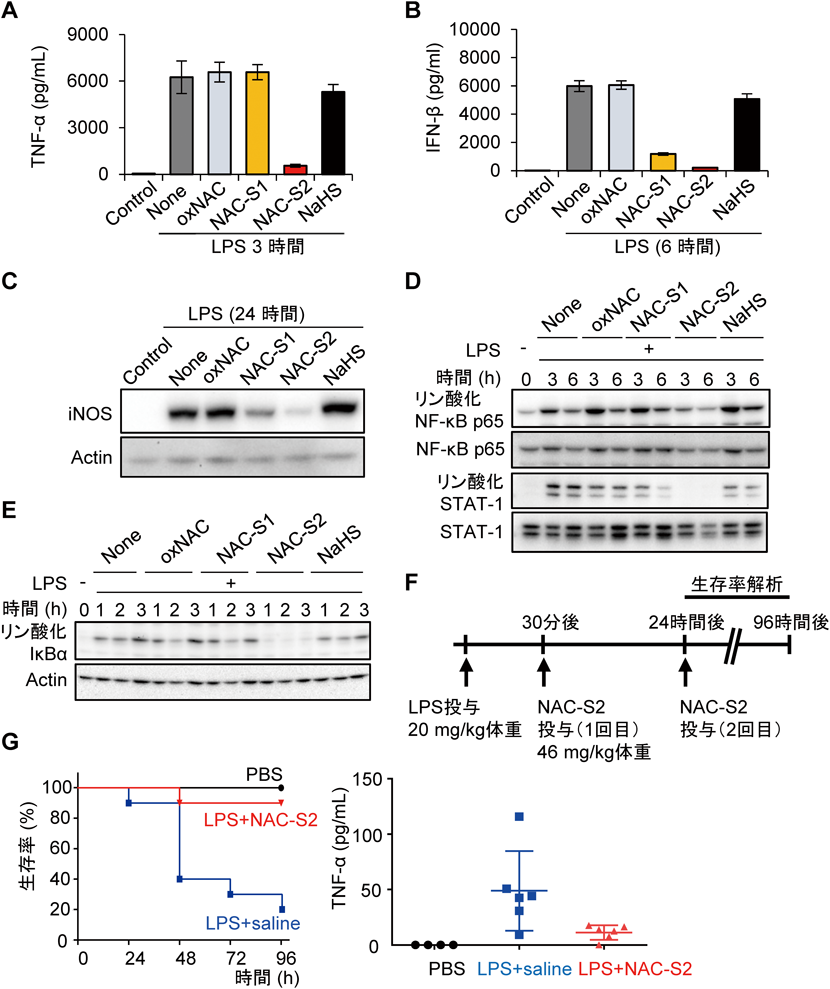
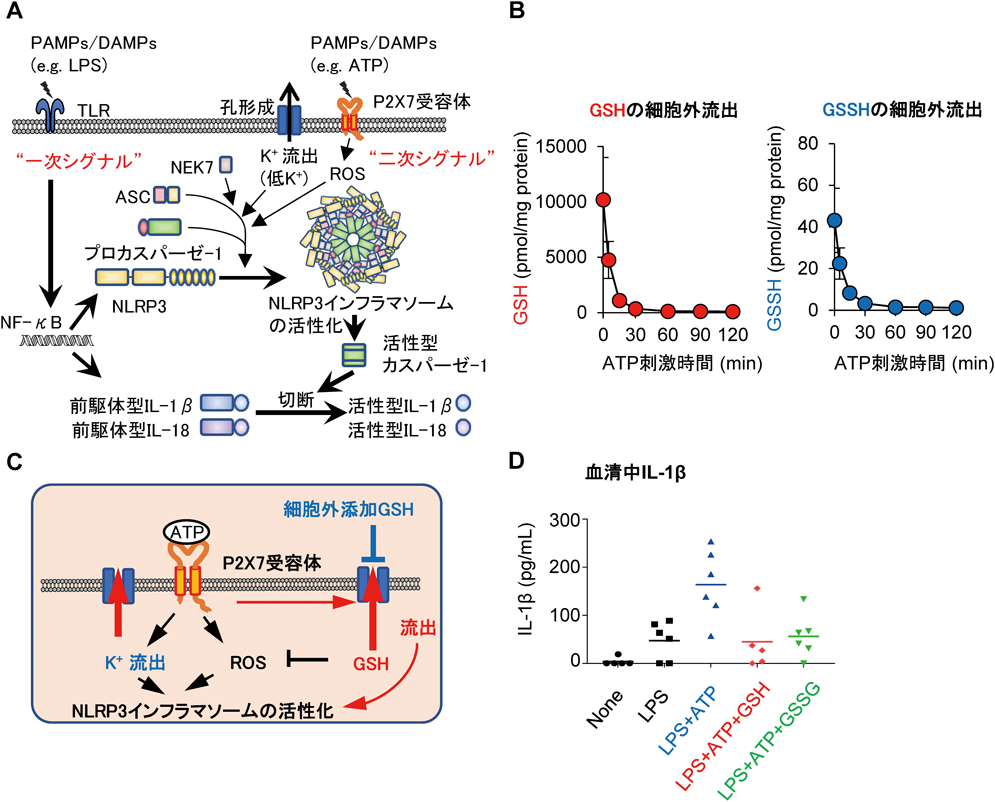
超硫黄分子の抗炎症作用Anti-inflammatory effects of supersulfides
熊本大学大学院生命科学研究部微生物学講座Department of Microbiology, Graduate School of Medical Sciences, Kumamoto University ◇ 〒860–8556 熊本市中央区本荘1–1–1 ◇ 1–1–1 Honjo, Chuo-ku, Kumamoto 860–8556, Japan
発行日:2021年10月25日Published: October 25, 2021