心臓には,心臓の収縮と弛緩を行う心筋細胞の他にもいくつかの細胞が存在している.線維芽細胞,内皮細胞,マクロファージ,肥満細胞,およびリンパ球などである.これら細胞の多くは均一な細胞群ではなく,いくつかの型があるのが特徴である.また,これらの細胞は,直接的あるいは放出した因子を介した相互作用により,健常時の恒常性維持に役立つとともに,損傷時の病態の進行や回復に寄与している4).各細胞について大まかに説明することで,これら細胞の果たす役割や機能,および細胞間の相互作用をイメージするときの助けとしたい.
1)線維芽細胞
線維芽細胞はECMの産生と分解によって細胞外環境を絶えず変化させていることが知られている3).線維芽細胞の数は少ないものの,心臓が損傷や慢性的な高血圧などのストレスにさらされると,線維芽細胞は,遊走活性が増加し増殖するようになるとともに筋線維芽細胞へ転換される.筋線維芽細胞は,コラーゲンやその他のECMを活発に産生する主要な細胞である.ここでは,特に断らない限り,ECMを産生しない細胞を線維芽細胞,産生する細胞を筋線維芽細胞とするこれまでの大まかな分類に従って述べていく.
2)内皮細胞
内皮細胞は,血管内腔に一層の状態で存在しており,血流の乱れや血圧の上昇などで傷を受けやすい.傷を受けた細胞は炎症を引き起こす.内皮細胞は,白血球が直接結合する接着分子を発現し,傷害後に生じる炎症に関与している.また,内皮細胞は一酸化窒素やプロスタグランジンなどの因子を放出し,血小板凝集の抑制や血管平滑筋の緊張を制御している5).心臓の血管に起きた炎症は,このような内皮細胞の創傷がトリガーとなるいくつかの過程を経て線維化を生じる.
3)マクロファージ
近年,心筋梗塞などにおいてマクロファージの関与が示されたことは,心疾患の原因として炎症が重要なことを示した注目すべき研究成果である6, 7).心臓のマクロファージには,常在性と浸潤性のマクロファージが存在する.常在性マクロファージとは,傷害を受けた心臓に浸潤してくるマクロファージとは異なり,心臓にあらかじめ存在しているマクロファージである.常在性マクロファージは,浸潤してくるマクロファージとは異なった性質を持ち,房室伝導を促進する作用を示すなど,心臓の恒常性の維持に重要な役割を果たしている8).傷害時には,常在性マクロファージは骨髄由来の単球から転換したマクロファージに置き換わる9).置き換わったマクロファージはサイトカインや成長因子などを分泌し,線維芽細胞を含む他の細胞に影響を与える.これらの作用は線維化の促進に関与している.また,マクロファージは,ECMを分解するプロテアーゼを分泌する.これにより,心臓のリモデリングと呼ばれる構造変化(心臓の病態が進行するにつれ,左室の拡大や収縮力の低下,発現しているイオンチャネルの変化,また線維化などが起こる現象)に関わっている.
4)マスト細胞
マスト細胞は傷害を受ける前の心臓にすでに存在している.マスト細胞は線維化領域で増殖し,線維芽細胞から筋線維芽細胞への転換を促進するサイトカインや成長因子を産生することで線維化に関わっていると考えられている10, 11).しかしながら,マスト細胞は線維化に関わる細胞として注目を受けているとはいえず,報告数もマクロファージや線維芽細胞に比べ少ない.
5)リンパ球
リンパ球は,心筋梗塞後の線維化に関与していることが報告されている12).Tリンパ球は,CD4+ T細胞とCD8+ T細胞という二つの大きなカテゴリーに分類される.いずれのT細胞にもsphingosine 1-phosphate receptor 1(S1P1)が発現しており,このS1P1を欠失させると,糖尿病時に生じる心臓の線維化が抑制された.これは,S1P1の欠失によって,糖尿病時に観察されるTGF-β発現量の増加が抑制されたことによっていた.興味深いことに,S1P1は血糖値が標準範囲にあるマウスでは線維化を抑制するように働いていた13).さらに,Tリンパ球は心筋細胞の生存に影響を与えるとともに,死んだ細胞がECMに置き換わることを促進する14).制御性T細胞(Treg)は,マクロファージの調節を行うTリンパ球である.Tregは圧負荷15)や心筋梗塞処置16)によって引き起こされる線維化を減弱させることが報告されているため,線維化病態を発症していない状態では,S1P1シグナルはこのTreg機能の調節を介し線維化を抑制し,線維化病態の発症時には,Th1やTh17などのT細胞機能を亢進させ,線維化を促進しているのかもしれない.
線維化で中心的な働きをする線維芽細胞および筋線維芽細胞について述べる.筋線維芽細胞は,ECMを産生する主要な細胞として認識されている.筋線維芽細胞は,心筋梗塞などのストレスを受けた後に線維芽細胞から転換され,欠落した領域の構造を維持する働きを持っている.これまで,心臓における線維芽細胞の数はかなり少ないと考えられてきた.しかし,最近のフローサイトメトリーを用いた研究では,線維芽細胞の心臓での割合は正常時でも約13%と,想定されていた以上の割合で存在していることが報告された17, 18).心臓が心筋梗塞や肥大などの傷害を受けると,線維芽細胞は筋線維芽細胞に転換され,ECMを産生するようになる.ここでは,線維芽細胞と筋線維芽細胞の特性について述べる.
1)線維芽細胞
組織特異的プロモーターの下流にマーカーとなる遺伝子を組み込んだマウスを用いると,特定の細胞がどのように変化していくのかを追跡することができる.これを系統追跡実験(lineage-tracing study)と呼んでいる.系統追跡実験の結果から,左心室および心室中隔に存在する線維芽細胞は,①内皮間葉転換(endothelial-mesenchymal transition:EndoMT)を介した心内膜細胞,②上皮間葉転換(epithelial-mesenchymal transition:EMT)を介した心外膜細胞に由来することが示された19).また,神経堤細胞からも少数の線維芽細胞が生成していた.ただし,成熟した内皮細胞,心外膜細胞および骨髄由来細胞は線維芽細胞に転換しない.マウスの心臓に負荷をかける処置として圧負荷や心筋梗塞処置がよく用いられる.圧負荷は,横行大動脈を狭窄する(閉塞ではない)処置を行って血管径を細くすることで,心臓がより強い力で血液を押し出さざるをえないようにする.これを圧負荷処置と呼んでおり,マウスの心臓に負荷をかける標準的な手法として広く用いられている.心筋梗塞処置は冠動脈を縛る(結紮する)処置である.異なった起源を持つ線維芽細胞から転換した筋線維芽細胞が,圧負荷処置後の心臓で固有の役割を担っているのか検討された.心外膜および心内膜の細胞を,特異的なプロモーター下にレポーター遺伝子を発現させ標識した.心内膜由来細胞[Tie2プロモーターの働きで緑色蛍光タンパク質(GFP)を発現する細胞]および心外膜由来線維芽細胞(Tbx18プロモーターの働きでGFPを発現する細胞)を標識し,圧負荷処置後,それぞれの細胞群を単離した.mRNAを分析すると,心内膜由来の線維芽細胞と心外膜由来の線維芽細胞の間で,mRNAは同様の発現パターンを示した19–21).さらに,心内膜由来と心外膜由来の細胞群は同程度の増殖活性を示した21).これらの結果は,線維芽細胞の起源が異なっていても機能に違いはないことを示している.ここで,線維芽細胞は通常のin vitroの培養条件では容易に筋線維芽細胞に転換するため,線維芽細胞をin vitroで解析することは現時点では困難であることをコメントさせていただく.
2)筋線維芽細胞
線維化で主要な働きをする筋線維芽細胞には,いくつかの特徴がある.①小胞体が広い範囲に存在している,②トロンボスポンジン,オステオポンチン,ペリオスチンなどのマトリセルラータンパク質(matricellular proteins)を合成している,③α-平滑筋アクチン(α-SMA)などの収縮性タンパク質を発現している,などいくつかの特徴がある22, 23).α-SMAは筋線維芽細胞の同定に広く用いられているタンパク質である.しかし,その発現は必ずしもα-SMA陽性細胞が筋線維芽細胞であることを意味しない.筋線維芽細胞への転換の初期段階では,筋線維芽細胞に似た細胞でα-SMAを発現していない細胞が検出される23).残念ながら,これまでのところ,筋線維芽細胞に特異的で信頼性の高いマーカータンパク質は報告されていない.
3)系統追跡実験による筋線維芽細胞の起源
筋線維芽細胞の起源は,細胞特異的プロモーター下にレポーター遺伝子を発現させた細胞を追跡することで検討された(系統追跡実験)19, 24).マーカータンパク質は発生や分化の途中で消失することがある.さらに,マーカータンパク質は解析しようとしている細胞だけでなく,機能的に無関係な細胞にも発現している場合もある.これに対し,遺伝子発現を指標にして系統を追跡する手法は,レポーター遺伝子がいったん発現すると,その細胞を発生の過程や他の細胞への転換などと関係なく継続的に観察できる.系統追跡実験では,目的とする細胞で特異的に働くプロモーターを用いてレポーター遺伝子を発現させる.これにより,プロモーターの活性がオフになった状態の細胞でも,過去にプロモーターが活性化された細胞ではレポーター遺伝子がすでに発現しているため,細胞を標識することができ,その後の変化を追跡することができる.
4)線維芽細胞の状態
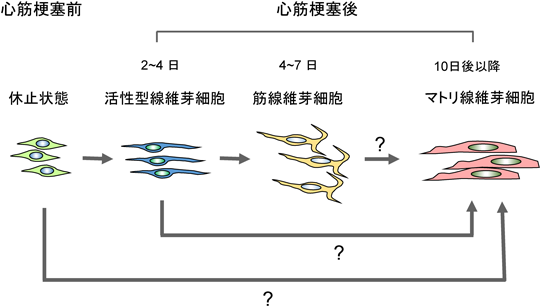
系統を追跡する技術を用いて,心筋梗塞後の線維芽細胞がどのように変化するのかを経時的に解析した報告がある24).その結果,線維芽細胞は四つの異なる状態をとりうることが明らかにされた.休止型線維芽細胞,活性型線維芽細胞,筋線維芽細胞およびマトリ線維芽細胞の4種である(図1).この報告では,休止状態にある線維芽細胞を,Tcf21プロモーターを利用して標識した.心筋梗塞後の線維芽細胞の増殖活性を5-エチニル-2′-デオキシウリジン(EdU)またはKi-67の標識によって測定したところ,増殖活性を有する線維芽細胞(活性型線維芽細胞)は,心筋梗塞の2~4日後に現れた.活性型線維芽細胞は,高い増殖活性および移動能力を有していた.筋線維芽細胞は,心筋梗塞の4~7日後に生じた24).筋線維芽細胞は,常在性線維芽細胞に由来していた.さらに,線維芽細胞は,心筋梗塞の10日後に新しいタイプの細胞(マトリ線維芽細胞)に転換した24).マトリ線維芽細胞がどの状態の線維芽細胞に由来するかは明らかではない.各状態の細胞で発現しているmRNAを分析すると,異なる状態にある細胞は異なるmRNAを発現していた.筋線維芽細胞はコラーゲンとα-SMAを産生していた.マトリ線維芽細胞は,瘢痕に局在し,腱や骨および軟骨で検出される骨関連遺伝子を発現していた.心臓におけるこれら骨関連遺伝子の生理学的意味は明らかではない.
5)シングルセルRNAシーケンシングによる線維芽細胞の分類
近年,筋線維芽細胞の不均一性が,シングルセルRNAシーケンシング(scRNA-seq)によって示された.scRNA-seqは急速に発展している技術で,それぞれの細胞が発現しているmRNA全体を質および量の側面から網羅的に調べる方法である25).細胞の遺伝子発現と細胞間の関係を包括的に特徴づけることができる.これまで,圧負荷によって心肥大が起きている状態の線維芽細胞は単一の細胞集団と考えられていた.しかし,11,492個の細胞のscRNA-seqにより,これらの細胞は単一の細胞集団ではなく,FB1からFB6の六つにグループ化された26).FB1は活性線維芽細胞に相当し,FB6はECMとペリオスチンを高度に発現する筋線維芽細胞に似た細胞である.各グループの細胞が線維化にどのように関与するか,FB1からFB6の細胞が,これまでに分類されてきた細胞に正確に対応するかは今後の課題として残されている.心筋細胞についてもscRNA-seq解析がなされている.ここでは線維芽細胞に関係のあるグループについてのみ記す.scRNA-seqの結果,心筋細胞は四つのグループ(FC1からFC4)に分けられた.このうち,FC3とFC4はカドヘリン5,フォンウィルブランド因子,ビメンチン,デコリンなどの内皮細胞や線維芽細胞のマーカータンパク質を発現していた.しかし,これらのグループは,転写因子Tcf21やPDGF受容体αなどの線維芽細胞を標識する標準的なマーカータンパク質のmRNAを発現していないことから,線維芽細胞由来ではないと考えられる.相関分析は,FC3およびFC4のグループが圧負荷処置後の心臓の病態と強く相関していた.FC3およびFC4のグループの持つ役割解析が待たれる.
この研究以外にも,scRNA-seqを利用した線維芽細胞の解析が報告されている.健常な心臓から線維芽細胞を単離しscRNA-seqを行ったところ,線維芽細胞の新しい状態が見いだされた27).この細胞集団は,線維芽細胞と免疫細胞の両方のマーカー分子を発現していた.ただし,健常時または傷害を受けたとき,これらの細胞が持つ機能や役割は明らかにされていない.また,系統追跡を利用してPDGF受容体αを発現する細胞を単離して,発現している各種の遺伝子を決定した報告もある28).その報告では,線維化促進性と抗線維化性の遺伝子をともに発現している新規な筋線維芽細胞のサブタイプが見いだされた.さらに,アンジオテンシンII処置後に存在する線維芽細胞の集団を解析した報告もある29).その報告によれば,α-SMAを発現する筋線維芽細胞が検出されず,マトリセルラータンパク質CILPとトロンボスポンジン4を発現する二つの線維芽細胞の集団が存在することが見いだされた.いくつかのscRNA-seqの研究に基づいて,線維芽細胞の状態に基づく新しい転換様式が提案された.この転換様式では,線維芽細胞には四つの異なる状態を想定している(図2)30).基底状態,増殖状態,活性化状態,終息状態の4種の状態である.基底状態では,発現している遺伝子の違いによって三つのサブセットにグループ化された.増殖状態の線維芽細胞は,四つの異なるサブセットにグループ化された.心臓が損傷を受けた後または老化の過程では,炎症性サイトカインが線維芽細胞を刺激し,線維芽細胞を活性化する.活性化状態にある一部の線維芽細胞は,増殖能力およびECMの産生能力を獲得する.活性化状態にある線維芽細胞の機能は,これまでにα-SMAの発現によって同定されてきた筋線維芽細胞とほぼ同じであった.興味深いことに,α-SMA陰性の細胞は血管新生の促進に関与している可能性が示された.増殖状態と活性化状態の線維芽細胞は互いに転換可能とされている.終息状態の線維芽細胞は表現型を,①基底状態の線維芽細胞へ復帰する,②アポトーシスによる消失を受ける,③老化した細胞へと変化させるに変化させた.これまでの研究によって,線維芽細胞が炎症を調節することが示されている.しかし,scRNA-seqでは,炎症を増強する活性を持つ線維芽細胞の集団は特定されていない.なお,各線維芽細胞の状態を特徴づける各種のマーカータンパク質も示されており,表1に示した.
表1 線維芽細胞の異なった状態で発現しマーカーとなるタンパク質| マーカータンパク質 | 基底状態 | 増殖型状態 | 活性化状態 | 終息状態 |
|---|
| コラーゲンタイプ1 (collagen type 1) | + | + | + | + |
| コラーゲンタイプ1受容体(DDR2) | + | | | |
| PDGF受容体α (PDGFR-α) | + | | | + |
| 転写因子21 (TCF21) | + | | | + |
| ペリオスチン(Periostin) | | + | + | |
| α-平滑筋細胞(α-SMA) | | | + (すべての細胞ではない) | |
| 軟骨中間層タンパク1, 2 (CILP1, 2) | | | + | + |
| 軟骨オリゴマーマトリックスタンパク質(COMP) | | | | + |
| トロンボスポンジン4 (thrombospondin 4) | | | + | |
| 血管内皮増殖因子-A (VEGF-A) | | | + | |
| 文献30を一部改変.+:発現あり,空欄:発現が認められていない,あるいは測定されていない.略号は次のとおりである.DDR2:discoidin domain receptor 2 (collagen type 1 receptor), PDGFR-α:platelet-derived growth factor receptor-α, TCF21:transcription factor 21, α-SMA:α-smooth muscle actin, CILP:cartilage intermediate layer protein, COMP:cartilage oligomeric matrix protein, VEGF-A:vascular endothelial growth factor A. |
これらの結果は,線維芽細胞の不均一性を示している.これまでのところ,各状態にある線維芽細胞間の相互作用や線維化への関与は明らかではない.系統追跡実験とscRNA-seqの結果に基づく線維芽細胞の分類には違いがみられ,この違いは,今後の研究の進展によって解決されていくと考えられる.
圧負荷や心筋梗塞の他に,心臓に線維化を引き起こす疾患として全身性硬化症が知られている.これまでのところ,全身性硬化症の線維芽細胞が,心筋梗塞時に誘導された細胞と同様に複数の状態を持つのかは明らかではない.
6)線維芽細胞,筋線維芽細胞のシグナルネットワーク
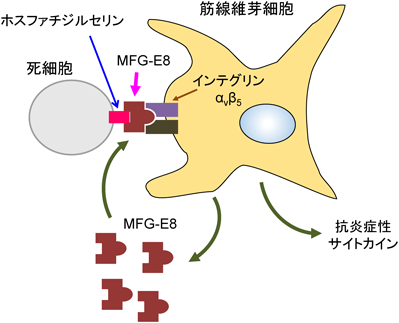
心臓の線維芽細胞は,心筋梗塞後2~3日に生じる炎症に関与することが報告されている31).線維芽細胞は,死につつある心筋細胞または死んだ心筋細胞から放出されるダメージ関連分子パターン(damage/danger-associated molecular pattern:DAMP)によっても活性化される.活性化された線維芽細胞は,炎症性サイトカイン,ケモカインおよびマトリックス分解タンパク質を産生する.これらの炎症性サイトカインは,線維芽細胞をさらに活性化して筋線維芽細胞への転換を促進させる.このように線維芽細胞と筋線維芽細胞は,損傷時には筋線維芽細胞への転換を促進する正のループを形成している.また,免疫細胞の損傷部位への動員は,炎症を負に制御するのにも役立っている.すなわち,活性化された線維芽細胞および筋線維芽細胞が分泌するケモカインは,損傷部位に免疫細胞を動員する.免疫細胞の動員は,マトリックスの破片の除去を刺激し創傷治癒を促進する作用もある.炎症の初期段階では免疫細胞を動員し,線維芽細胞から筋線維芽細胞への転換を促進させる.筋線維芽細胞は,免疫細胞とともに死細胞の除去を促進する.死細胞の除去は,抗炎症性サイトカインの分泌を促進することから,炎症を減弱させるように働く.同時に,筋線維芽細胞によるECMの産生が増加し,損傷部位の構造維持に働く.
7)自己免疫性心筋炎における筋線維芽細胞の起源
自己免疫性心筋炎は,CD4+ T細胞を介した炎症性の心筋炎である.実験的に自己免疫性心筋炎を起こすと,心臓に浸潤してくるCD133+前駆細胞が,TGF-βの助けを借りて筋線維芽細胞様の細胞に転換されることが報告されている32).筋線維芽細胞の起源となる細胞は,疾患の種類によって異なっている可能性がある.
5. 筋線維芽細胞への転換を制御するシグナル伝達経路
線維芽細胞から筋線維芽細胞への転換を制御するシグナル伝達として多くのシグナル伝達経路やシグナル分子が知られている.ここではそれらの中からいくつかを紹介する.
1)炎症に関わる分子
DAMPsは損傷を受けた細胞から放出され,炎症を引き起こす因子の総称である37).損傷部位に動員される炎症細胞は,TGF-βを含むサイトカインを放出する.TGF-βは,線維芽細胞から筋線維芽細胞への転換を強く引き起こす38).また,DAMPsやTGF-β以外のサイトカインも,線維芽細胞から筋線維芽細胞への転換を促進する.したがって,炎症の阻害は筋線維芽細胞の出現を抑制することにつながり,線維化を減弱させることが期待できる.
心筋梗塞後の早い時期,損傷部位へ動員されるのは好中球である39, 40).ロイコトリエンB4は,好中球のロイコトリエンB4受容体(BLT1)に結合し,好中球の損傷部位への移動を強く促進する.BLT1遺伝子の欠損,あるいはBLT1拮抗薬の投与で好中球の動員を阻害させたりすると,炎症を減弱させることができ,それとともに線維化が減少した41, 42).さらに,心機能の低下が抑制されたことから,炎症が線維化と心機能の低下に結びついていることを示している.
2)インターロイキン
インターロイキン(IL)は,炎症性サイトカインのメンバーである.複数のIL受容体が線維芽細胞に発現しており,線維芽細胞の状態と機能を調節している.線維芽細胞に対する炎症性ILの役割は,IL受容体を線維芽細胞から特異的に欠失させたマウスの解析によって検討された.IL11受容体またはIL17受容体遺伝子のノックアウトマウスは,損傷によって引き起こされる線維化および心機能の低下が減弱していた43, 44).さらに,IL受容体の遺伝子を線維芽細胞特異的に欠失させたマウスでは,損傷領域への免疫細胞の浸潤や活性が低下していた.これは,線維芽細胞がILシグナル伝達を介して,損傷によって引き起こされる炎症に重要な役割を果たしていることを示している.
3)マクロファージ
心筋梗塞後,単球が骨髄から動員され,損傷部位でマクロファージに転換する45).マクロファージは,クロドロネート・リポソームでマウスを処置することによって枯渇させることができる46).マクロファージが枯渇したマウスは,線維化の減少と心機能の改善を示した.これは,心筋梗塞後の炎症性シグナル伝達の阻害が,線維芽細胞から筋線維芽細胞への転換を阻害することで,線維化を減少させることを示している.
4)TGF-βシグナル
TGF-βで細胞を刺激すると,標準的なSmad2/3と非標準的なMAPキナーゼが活性化する28).TGF-βシグナル伝達の線維化における役割は,線維芽細胞特異的にTGF-β受容体1および2遺伝子を欠失させたマウス,またはTGF-β受容体の下流で活性化される転写因子Smad3遺伝子を欠失させたマウスを解析することで得られた.ノックアウトマウスの作製に用いたペリオスチンのプロモーターは,常在性の線維芽細胞で働いておらず,筋線維芽細胞への転換とともに活性化される性質を持っている.これらマウスでは,圧負荷によって引き起こされる線維化が阻害された47).興味深いことに,線維芽細胞におけるTGF-β受容体1および2遺伝子の欠失が,圧負荷による心肥大も抑制した47, 48).心肥大時には心筋細胞の大きさが増大する.したがって,筋線維芽細胞との直接的な細胞間相互作用により,あるいは筋線維芽細胞が分泌する因子を介した間接的作用により,筋線維芽細胞が心筋細胞に影響している可能性を示している.
5)リゾホスファチジン酸シグナルとMRTF-SRFシグナル伝達
リゾホスファチジン酸(LPA)刺激は,線維芽細胞から筋線維芽細胞への転換を引き起こす36, 37).LPAはGタンパク質共役受容体(GPCR)に結合して作用を発揮する49).in vitroでは,LPA刺激はミオカルディン関連転写因子–血清応答因子(myocardin-related transcription factor-Serum response factor:MRTF-SRF)経路の活性化によって線維化を引き起こした.Rho(Ras homolog gene family)によって調節されるアクチンのオリゴマー化は,Rho関連キナーゼ(Rho-associated kinase:ROCK)によるアクチンのリン酸化によって調節されている.ROCKがアクチンをリン酸化すると,単量体型のアクチンが増加する.単量体型アクチンはSRFを阻害する活性を持たない.阻害から解放されたSRFはMRTFとともに働き,線維化を促進する遺伝子を含むさまざまな遺伝子の転写を活性化する50).Rhoは,GPCRやTGF-β受容体などのさまざまな受容体の下流で活性化されるシグナル伝達分子である.心臓にはRhoを活性化する数多くの受容体が発現している.したがって,Rho-ROCK経路の阻害剤は,多くの受容体を同時に阻害するよりも効率的に線維芽細胞の活性化と線維化を抑制する可能性がある.
臨床への適用を考えると,低分子化合物の合成は欠かせない.MRTF-SRFシグナル伝達経路の阻害剤としてCCG-203971が報告されている51).CCG-203971は,新生仔ラットより単離された線維芽細胞を用いて,YAP(Yes-associated protein)-MRTFシグナル伝達経路による線維化応答を阻害した52).YAPはTGF-β刺激によって活性化される転写因子の一つである.核内へ移行したYAPはMRTF発現を促進することで,線維化を引き起こす.これまでのところ,個体レベルでCCG-203971が心臓の線維化に効果を示したという報告はなされていない.YAPについては6節3項のHippoシグナルのところでも述べる.
6)Gタンパク質共役型受容体キナーゼ2(GRK2)
GRK2は,アゴニストが結合したGPCRをリン酸化することにより,GPCRの活性を調節する48).α-ミオシン重鎖(α-myosin heavy chain:α-MHC)プロモーター下にCreを発現させたGRK2の心筋細胞特異的なノックアウトマウスは,心筋梗塞による線維化および心機能の低下が減弱していた53).また,このマウスは心筋梗塞による心不全の発症が減少した.GRK2の線維芽細胞での役割も検討されている.コラーゲン1α2プロモーターを使用したGRK2の線維芽細胞特異的なノックアウトマウスでは,TNF-αの分泌が減少し,虚血再灌流後の線維化を促進する因子の遺伝子発現が抑制された54).また,心機能の低下も改善された.これらの結果は,心筋細胞および線維芽細胞でのGRK2の阻害が,心臓をストレスから保護することを示している.
7)transient receptor potential channel canonical(TRPCチャネル)
TRPCファミリーは,電位に依存しない陽イオンチャネルであり,開いている状態ではCa2+やNa+などを濃度勾配に従って透過させる.TRPCファミリーのメンバーであるTRPC3および6(TRPC3/6)は,アンジオテンシンIIで刺激されたときの心筋細胞での肥大応答を仲介していることが報告された55).TRPC3/6はジアシルグリセロール(DG)によって活性化されチャネルを開く.ラット新生仔心室筋細胞では,アンジオテンシンII刺激は肥大応答を引き起こす.アンジオテンシンIIはGq共役型のアンジオテンシンII受容体1型に結合し,ホスホリパーゼCを活性化する.これによりイノシトール1,4,5-三リン酸(IP3)とDGが産生される.DGはTRPC3/6を介して脱分極を引き起こし,最終的に細胞内Ca2+を増加させる.増加したCa2+によりnuclear factor of activated T-cells(NFAT:T細胞に関連する核因子)の核への移行が促進され,NFATにより心肥大応答が引き起こされる.TRPCを介した細胞内Ca2+の増加は,線維芽細胞から筋線維芽細胞への転換にも重要な役割を果たしていることが報告された56).線維化は筋線維芽細胞に依存して生じる.TRPC6は線維化を引き起こすのに十分な細胞シグナル伝達を活性化できる.TGF-β刺激によってTRPC6の発現上昇が引き起こされ,TRPC6を介した応答は,p38-MAPKを介したシグナル伝達の遮断によって阻害された57).さらに,TRPC6をノックアウトしたマウスから単離した線維芽細胞では,細胞をアンジオテンシンIIやTGF-βで刺激してもα-SMA発現上昇が抑制され,線維芽細胞から筋線維芽細胞への転換が阻害された.これらの結果は,TRPC6-Ca2+シグナル伝達が線維芽細胞から筋線維芽細胞の転換に大きな役割を果たしていることを示している.
8)一次繊毛の役割
一次繊毛は細胞の表面から外側に伸びている1本の構造体であり,哺乳動物のほぼすべての細胞に存在している.心臓を含む組織の発生,成長に関わっている.線維芽細胞の一次繊毛が,TGF-β刺激による線維芽細胞から筋線維芽細胞への転換に必要であることが見いだされた58).ポリシスチン-1は,細胞増殖,細胞移動および他の細胞と相互作用する際の調節因子として知られているタンパク質であり,一次繊毛に発現している.このポリシスチン-1は一次繊毛の細胞構造を維持するために必須である.ポリシスチン-1遺伝子の欠失によって線維芽細胞の一次繊毛が破壊されると,TGF-β-Smadシグナル伝達によって引き起こされるECMタンパク質の産生および筋線維芽細胞への転換が阻害される.ポリシスチン-1遺伝子を欠失させたマウスでは,心筋梗塞後のリモデリングが増強されることから58),一次繊毛は線維芽細胞の筋線維芽細胞への転換および線維化において重要な役割を果たしている.
9)熱ショックタンパク質(Hsp)
Hspは,線維芽細胞から筋線維芽細胞への転換に必要なシャペロン分子である.さまざまなHspタンパク質の中で,Hsp47はコラーゲン特異的シャペロンとして知られている.線維芽細胞のHsp47遺伝子を特異的に欠失させたマウスでは,圧負荷後の線維化が有意に減少し,拡張機能の低下が減弱した59).しかし,これらのマウスでコラーゲン産生が減少すると,心筋梗塞処置により欠落した細胞を補う瘢痕形成が不十分となり致死率が増加した.
これらの報告は,線維化に関与するさまざまなシグナル伝達分子の発現および活性の制御が,線維芽細胞の状態の調節を介した線維化の阻害に結びつくことを示している.
10)アンジオテンシンII
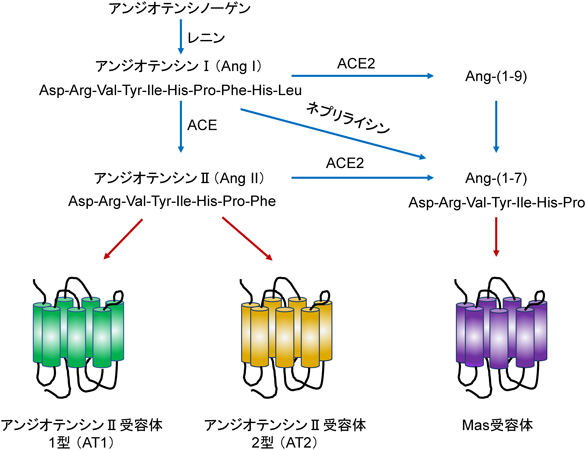
アンジオテンシンII(Ang II)は,循環系の調節できわめて重要な分子であり,その作用を減弱させる薬剤は,心不全や高血圧などの薬物治療で広く使われている.Ang IIの産生へと至る経路は,レニン-アンジオテンシン系と呼ばれている(図3).この経路は,Ang IIによって発現が調節され体液量の調節に働くアルドステロンを含めたレニン–アンジオテンシン–アルドステロン系とも呼ばれる.
アンジオテンシノーゲンにレニンまたはキマーゼが作用するとアンジオテンシンI(Ang I)が生成する.Ang Iはアンジオテンシン変換酵素(ACE)の作用によってAng IIを生成する.Ang IIは,アンジオテンシン1型(AT1)および2型(AT2)受容体に結合して作用を発揮する.Ang IIは血圧調節など循環系の制御に重要な役割を果たしており,ACE阻害薬とAT1受容体拮抗薬は血圧の低下や心不全の治療などに用いられている.生体内の循環系に対する作用はAT1受容体を介して発現する.AT1受容体はGPCRファミリーのメンバーである.Gタンパク質はGs, Gi, GqおよびG12の4種のサブファミリーに分けられる60).Gsはアデニル酸シクラーゼを活性化しcAMP産生を増加させる.Giはアデニル酸シクラーゼを阻害するとともにK+チャネルを活性化し過分極を引き起こす.GqはホスホリパーゼCを活性化し,IP3とDGを生成する.G12はRhoを活性化する.多くのGqに共役するGPCRはG12サブファミリーとも共役する.また,GqがG12とは独立したシグナリング分子を介してRhoを活性化する経路も存在する.AT1受容体の活性化は,Gqを介してCa2+の増加とRhoの活性化を引き起こし,細胞応答を引き起こす.心筋細胞で上昇したCa2+は,Ca2+感受性プロテインキナーゼとCa2+調節転写因子を活性化し,心肥大を引き起こすように働く.一方,線維芽細胞でのRhoの活性化はMRTF-SRFシグナル伝達経路を刺激し線維化を促進する.Ang IやAng IIは,ネプリライシンまたはACE2の作用により,さらにアンジオテンシン-(1-7)[Ang-(1-7)]に代謝される.Ang-(1-7)は,GPCRであるMas受容体を介してAng IIの作用に拮抗する61).Ang-(1-7)の作用はAng IIを介する作用に拮抗することから,創薬のターゲット分子として期待できる.しかし,Mas受容体は循環系以外の組織にも発現し生理応答の制御に関わっている.したがって,Mas受容体を活性化する化合物の線維化治療への適用は,投与期間が長期になることと相まって,困難が予想される.
11)活性酸素種(reactive oxygen species:ROS)
AT1受容体刺激は,Gqを介してNADPHオキシダーゼ(NOX)を活性化し,ROSを産生させる.ROSは用語であり,分子を表しているわけではではない.ROSの標的分子は明確に特定されていないものの,さまざまなタンパク質のシステインのチオール基(スルフヒドリル基とも呼ぶ)の修飾によって,その効果を発揮すると考えられている.さまざまなROSの中で,過酸化水素(H2O2)はAT1受容体のみならず他の受容体刺激によっても生じ,安定性や反応性などからさまざまなタンパク質のチオール基修飾における主要なROSとして認識されている62, 63).ROSの線維化への関与については,8節「線維化の治療法」の4項「アクアポリンの阻害」であらためて述べる.
12)筋線維芽細胞の細胞数の制御
アポトーシスはプログラムされた細胞死の一形態であり,高度に調節されたプロセスである64).アポトーシスには,内因性経路(ミトコンドリア経路)と外因性経路の2種類がある.内因性経路では,細胞ストレス,DNAの損傷,発生の開始あるいは生存因子の除去によってアポトーシス経路が活性化され細胞は死滅する.内因性経路では,シトクロムcはBaxタンパク質によってミトコンドリアから放出される.放出されたシトクロムcは,アポトーシスプロテアーゼ活性化因子-1(Apaf-1)と複合体を形成する.この複合体はプロカスパーゼを切断して,活性型カスパーゼ-9を生成させる.次に,カスパーゼ-9は,細胞内のさまざまなタンパク質を切断するカスパーゼ-3に作用する.一方,外因性経路では,細胞外のシグナル分子が細胞死受容体に結合してアポトーシス経路を活性化することで細胞を死滅させる.外因性経路では,TNF-α受容体などの細胞死受容体が経路を刺激し,TNF受容体関連因子(TRAF)などの中間タンパク質を介してカスパーゼを活性化する.その結果,カスパーゼ-9および-3が活性化され,さまざまなタンパク質が分解される.アポトーシスを促進させるシグナルに拮抗する分子群としてBcl-2ファミリーが存在している.アポトーシス促進性(Bax)タンパク質と抗アポトーシス性(Bcl-2)タンパク質の比率によって,細胞の生存と死が決まる.次に,創薬との関連が注目されているBcl-2ファミリーについて述べる.
13)Bcl-2ファミリー
Bcl-2ファミリーはBcl-2相同(Bcl-2 homology:BH)ドメインを持つタンパク質群であり,ミトコンドリアの膜透過性を制御することによりアポトーシスを制御している.Bcl-2ファミリーのメンバーは,BHドメインと呼ばれる四つのドメイン(BH1~BH4)を一つ以上持っているのが特徴である.構造と機能に基づいて,Bcl-2様,Bax様,BH3-onlyの三つのグループに分けられる.Bcl-2様タンパク質は抗アポトーシス活性を持っている65).Bax様およびBH3-onlyタンパク質はアポトーシスを促進する.アポトーシスを促進するBax様およびBH3-onlyタンパク質は,Bcl-2に存在する疎水性BH3結合ポケットに結合してヘテロ二量体を形成し(BH3類似体),Bcl-2による抗アポトーシス活性を阻害する.これによりアポトーシスを促進する66).
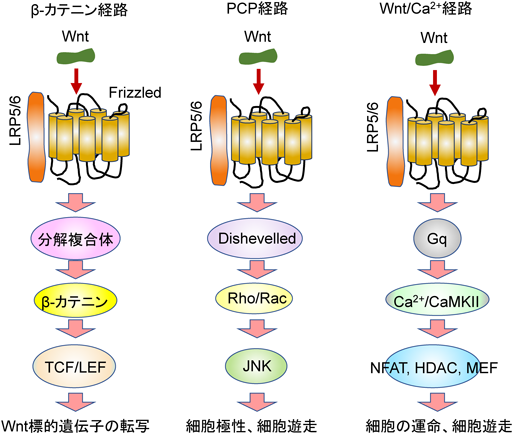
14)Wntシグナル伝達
Wntシグナル伝達は,標準的なシグナル伝達と非標準的なシグナル伝達に分類される.β-カテニンは,標準的なWntシグナル伝達において重要な役割を果たしている(図4)67).β-カテニンは,転写因子TCF(T-cell factor:T細胞因子)/LEF(lymphoid enhancer factor:リンパ系エンハンサー因子)の転写活性を上昇させることで,細胞周期や接着などのさまざまな応答を調節している.β-カテニン遺伝子を線維芽細胞で欠失させると,筋線維芽細胞によるECMタンパク質の産生が減少した.その結果,線維化が減弱し心機能も改善された68).
非標準的なシグナル伝達は,Wnt/PCP(planar-cell-polarity:平面細胞極性)経路とWnt/Ca2+経路に分かれる.二つの非標準的なシグナル伝達経路は,β-カテニン-TCF/LEFシグナル経路とは独立している.Wnt/PCPシグナル伝達は,Wnt5aやWnt11などのWntタンパク質が,Fzd(Frizzled)受容体の一つおよび受容体チロシンキナーゼ様オーファン受容体2(ROR2)に結合することによって開始される69, 70).これら受容体の刺激は,細胞膜にDvl(dishevelled)を移動させRhoとRacの活性化を促進させる.活性化されたRhoは,Rho関連キナーゼ(ROCK)とc-Jun-N末端キナーゼ(c-Jun-N-terminal kinase:JNK)を活性化し,各種遺伝子の発現を増加させる.非標準的なシグナル伝達のもう一つのWnt/Ca2+経路は,WntがFzd受容体に結合することによって活性化される.刺激によってGqが活性化され,細胞内貯蔵部位からのCa2+放出が引き起こされる.Ca2+の上昇は,カルモジュリン依存性キナーゼII(CaMKII)と転写因子NFAT, HDAC4(histone deacetylase 4:ヒストンデアセチラーゼ4)とMEF(myocyte enhancer factor:筋細胞エンハンサー因子)を活性化する.Wntシグナル伝達は,線維化の引き金の一つであると報告されている.Wntシグナル伝達はヒト心血管疾患の病態時でも活性化されており71),Wntシグナル伝達を調節する化合物が動物モデルで有望な結果を示している72).しかし,化合物の臨床応用はこれまで報告されていない.
線維芽細胞は心筋細胞の間に存在している.心臓が心筋梗塞や圧負荷などのストレスにさらされると,線維芽細胞は筋線維芽細胞に転換し,コラーゲンなどのECMを産生するようになる.損傷部位で生じた筋線維芽細胞などは,活発に増殖するとともに損傷部位から遊走する活性も示す.通常の培養条件では,線維芽細胞は単離して培養するのみで容易に性質が変化するため,これまでin vitroでの詳細な解析は困難であった.ここでは,新しい培養の試み,線維芽細胞の性質を制御する因子やシグナリングなどについて紹介する.
1)3次元培養システムを用いた解析
線維芽細胞のいくつかの状態を反映するin vitro 3次元培養システムが開発された73).線維芽細胞を単離して2次元(2D)または3次元(3D)で培養すると,線維芽細胞の性状が変化した.2D培養には標準のポリスチレンでコートした培養プレート(コラーゲンやその他のECMでコーティングされていない)を使用し,3D培養では超低接着プレート(ECMでコーティング)を使用した.3D構造プレートで培養された線維芽細胞は球形の塊を形成した.形態変化は,細胞外の張力または硬さ(剛性)には依存していなかった.しかし,3D培養の線維芽細胞とリモデリングを起こした心臓(3週間のイソプレナリン処置)の間には発現遺伝子の相関関係がみられた.3D培養の線維芽細胞ではα-SMAの発現は減少していた.筋線維芽細胞のマーカーであるα-SMAの発現が低下しているため,3D培養で得られた線維芽細胞が従来の筋線維芽細胞に相当するとはいえない.しかし,3D培養の線維芽細胞で得られたmRNAの発現パターンは,マトリ線維芽細胞の発現パターンと類似していた74).したがって,3D培養で得られた線維芽細胞を分析することで,マトリ線維芽細胞の機能やどのような変化をたどるのかなどを明らかにできる可能性がある.さらに,3D培養の線維芽細胞は,細胞を2D培養に移すことにより,形態と発現遺伝子を可逆的に変化させた.線維芽細胞から筋線維芽細胞への転換メカニズムとそれぞれの細胞の機能を解析するにはin vitroの測定系が必要である.簡便な培養条件の交換による線維芽細胞の形態変化は,線維芽細胞の複雑な挙動を分析するための有用な手法となる可能性がある.
2)ECMの硬さ
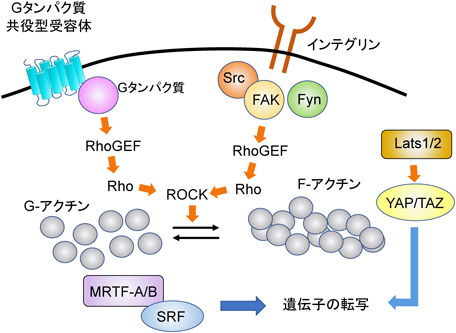
線維芽細胞の筋線維芽細胞への転換は,ECMの硬さによっても調節されている75).線維芽細胞の周りの硬さは,線維化が進行するにつれて上昇する.細胞周りの硬さは,インテグリン受容体とアクチン細胞骨格によって感知され,p38-MAPKの核への移行を促進させる.その後,移行したp38-MAPKがリモデリングに関わるさまざまな遺伝子の発現を増加させる76).また,インテグリン-アクチン細胞骨格からなるシグナル伝達複合体は,接着斑キナーゼ(focal adhesion kinase:FAK),Src, Fynなどのチロシンキナーゼも活性化する.これらのキナーゼは,Rhoのグアニンヌクレオチド交換因子(RhoGEF)を介してRhoのGDP-GTP交換を促進する.RhoはROCKを介してアクチンの重合を促進させ,MRTFとSRFの協調作用による線維化に関連する遺伝子の転写を増加させる77).
3)Hippoシグナル伝達経路による制御
Hippoシグナル伝達経路(Hippoシグナルと略す)は,細胞増殖とアポトーシスの調節を通じて器官のサイズを制御するシグナル伝達経路である.心臓でのHippoシグナルは,心筋細胞の増殖を阻害することが知られている78).YAPは,Hippoシグナル伝達の転写コアクチベーターであり,large tumor suppressor kinase 1(Lats1)およびLats2(図5)によって負に調節されている.Hippoシグナルで働いているシグナリング分子の線維芽細胞における役割が解析されている.Col1α1プロモーターを用いて線維芽細胞からYAPを欠失させると,心筋梗塞後のコラーゲンの蓄積および線維芽細胞の増殖や活性化は減少した52).YAPをノックダウンした線維芽細胞では,アンジオテンシンII刺激による線維化応答は減弱していた79).さらに,MRTF-Aの発現は,YAPをノックダウンさせた細胞では減少した.MRTF-Aの発現はYAPによって調節されているといえる.また,これらの結果は,YAP-MRTF-Aシグナル伝達経路が,心筋梗塞や神経体液性因子に応答した線維芽細胞から筋線維芽細胞への転換に,重要な役割を果たしていることを示している.興味深いことに,YAPの心筋細胞特異的な欠失は肥大を減少させ,線維化を有意に増加させた80).このことは,心筋細胞と線維芽細胞でYAPが異なる役割を持っていることを示している.
Lats1とLats2はYAPをリン酸化し,YAPを介したHippoシグナル伝達の転写活性を阻害する81).Lats1とLats2を,Tcf21プロモーターを用いてノックアウトさせたマウスでは,YAPの核内移行が増加するとともに,自発的な筋線維芽細胞への転換を引き起こした82).さらに,Lats1およびLats2のノックアウトマウスは,健常時と心筋梗塞後の両方で線維化の増加を示した.これは,YAPが線維化を引き起こす因子として働くこと,さらにLats1/2はYAPの阻害を介して線維化に関わることを示している.
Lats1およびLats2の機能は,Rhoによって制御されるアクチン細胞骨格によって調節されている.Rhoは,MRTF-AおよびMRTF-Bを介して線維化にも関与している.MRTF-Aおよび-MRTF-Bは,SRFが血清応答エレメント(serum response element)として知られるプロモーター配列に結合するのを助ける.その結果,Rhoの下流で転写が活性化され,線維化を引き起こす.このように,Rho, Lats1/2, YAP, MRTF-A/BおよびRhoは,複雑なシグナルネットワークを形成し線維芽細胞を介した応答を制御している.
線維化は,体のさまざまな組織で生じ,適切な治療法が存在しないため,アンメットニーズの高い病態である.これまでに薬効を示す薬剤がほとんどないといってもよい状況である.ここでは動物モデルで有効性を示し,患者でも効果が期待できそうな化合物を紹介する.
1)TGF-βシグナリングに作用する薬剤
TGF-βは線維化を強力に引き起こすため,TGF-βシグナル伝達経路の阻害剤が抗線維化薬として有効性を示すことが期待されてきた38).TGF-βシグナル伝達阻害剤をブレオマイシンによる肺線維化およびアルコールによる肝線維化の治療に使用した報告がある86).しかし,心臓の線維化にTGF-βシグナル伝達阻害剤が有効性を示したという報告はなされていない.
特発性肺線維化の治療薬として,ピルフェニドンとニンテダニブが承認されている.ピルフェニドンはTGF-β産生を阻害する.一方,ニンテダニブはTGF-βシグナルの阻害作用ではなく血管内皮成長因子受容体,線維芽細胞成長因子受容体,血小板由来成長因子受容体の各受容体のチロシンキナーゼ活性を阻害する87).しかし,これら二つの薬剤を含むシグナル伝達阻害剤は,心臓の線維化に対して有益な効果を示さなかった88).抗線維化薬の標的分子は,心臓やその他の組織において修復プロセスなどの重要な細胞応答に関与している.したがって,抗線維化薬による長期にわたる治療は,望ましくない影響(重篤な副作用)を引き起こす可能性がある.事実,TGF-βシグナル伝達阻害剤には,副作用のために開発が断念されているものがある89).
2)GRK2を阻害する薬剤
GRK2は,アゴニストに結合したGPCRをリン酸化するキナーゼ(G protein-coupled receptor kinase:GRK)ファミリーのメンバーである.化合物のスクリーニングから,パロキセチンがGRK2阻害剤として同定された.パロキセチンはGRKファミリーに属する他のメンバーを阻害しなかった.パロキセチンは選択的セロトニン再取り込み阻害薬(SSRI)として知られている薬剤である.パロキセチンの投与により,心筋梗塞後の心不全および線維化が減弱した90).パロキセチンの作用がSSRIによる効果ではないことを確認するため,パロキセチンとは異なるSSRIであるフルボキサミンの作用が検討された.フルボキサミンはGRK2を阻害せず,心不全の症状を軽減しなかった66, 90).線維化に対する効果は測定していないものの,線維化は多くの場合,心不全に付随して起こるため,線維化は抑制されていないと思われる.しかし,パロキセチンのGRK2に対する親和性は,GRK2のリン酸化活性を抑制する作用から評価したとき,EC50値がμM以上と低いことから91),高親和性のGRK2選択的阻害剤の開発が待たれている.
3)アポトーシスを促進する薬剤
ナビトクラックス(ABT-263としても知られている)はBH3類似体であり,Bcl-2に結合することでアポトーシスを促進させる92).強皮症は心臓を含む全身にコラーゲンが過剰に沈着する病気である.ナビトクラックスは,強皮症の患者から分離された筋線維芽細胞のアポトーシスを促進した93).筋線維芽細胞のアポトーシスを増強すると線維化からの回復が効果的に進行するため,ナビトクラックスは線維化を治療するための有望な薬剤となる可能性がある.筋線維芽細胞のアポトーシスの増強は,線維化の創薬ターゲットになりうる.Bcl-2に対して優れた選択性を有するベネトクラクスは,現在,抗がん剤として臨床試験が行われている94).将来的には,ベネトクラクスが心臓の線維化の治療にも適用される可能性がある.
4)アクアポリンの阻害
アクアポリンはH2O2を細胞外から細胞内に移動させる働きを持っている95).マウスの心臓には,アクアポリンの数種のアイソフォームが発現している.アクアポリンを通って細胞内に入ったH2O2はタンパク質を酸化修飾し,その機能を変化させることで有害な作用を起こす.H2O2はNADPHオキシダーゼによって産生されるスーパーオキシドアニオン(O2−)から細胞外で生成する96, 97).心臓でH2O2を移動させるアクアポリン1のノックアウトマウスが,圧負荷やAng IIの持続投与による心肥大と線維化の抑制を示したことから,化合物による効果が検討された.アクアポリン1の阻害薬であるバコパシド(Bacopaside)は,in vitroでのAng IIをはじめフェニレフリン,イソプロテレノール刺激による肥大応答を抑制した.バコパシドそのものは動物で効果を確認するには費用がかかりすぎるため,アーユルヴェーダ伝統医学で不安やストレスを軽減するために用いられているバコパシドを含む抽出物を用いて検討した.抽出物を圧負荷やAng IIの持続投与を施したマウスに投与すると,心肥大と線維化が減弱した98).これらの結果は,アクアポリンが線維化および肥大を治療するための有望な標的であることを示している.
漢方の五苓散は,利尿作用を期待して患者に用いられている.五苓散は後ろ向きの試験ながら,慢性腎臓病合併性心不全患者に対する標準治療に加えた投与で,安全でかつ有効なことが示されている99).しかし,利尿効果を持つ薬剤フロセミドは絶水状態でも浮腫状態でもどちらも尿量が増えるのに対し,五苓散は浮腫状態でのみ尿量が増えることが示されている.すなわち,心不全への五苓散の効果は利尿作用では説明できない.五苓散がアクアポリンを阻害することから100),バコパシドと同様に心不全や線維化に効果を示すことが期待される.なお,アクアポリン阻害活性を持つ五苓散の成分は,金属のマンガンあるいはマンガンを含む化合物と報告されている.
5)CAR T細胞
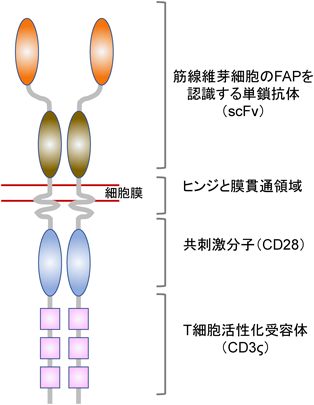
心臓の線維化を治療標的にした免疫学的方法が報告された101).キメラ抗原受容体(Chimeric antigen receptor:CAR)T細胞は,筋線維芽細胞を選択的に認識し,筋線維芽細胞の消失を引き起こすことで心機能を改善させた.CARは,抗原を認識する単鎖Fvフラグメント(single chain Fv fragment:ScFv),ヒンジと膜貫通ドメイン,共刺激分子(co-stimulation receptor:CD28)の細胞内ドメインおよびT細胞活性化受容体(CD3ζ)から構成されている(図7).CARが筋線維芽細胞特異的タンパク質[この場合はマウス線維芽細胞活性化タンパク質(fibroblast activation protein:FAP)]に結合した後,CAR T細胞は筋線維芽細胞の細胞死を誘導させ,その数を減少させた.CAR T細胞はすでにがん治療に使用されており,有益な効果を示すと報告されている.筋線維芽細胞の減少が線維化の治療に役立つことが,次の実験によって裏づけられている.
ヒトのジフテリア毒素受容体を筋線維芽細胞に特異的に発現するマウスを作製する.マウスの受容体はヒトのジフテリア毒素と結合しないため,ヒトのジフテリア毒素を投与することでヒトの受容体を発現している細胞のみを選択的に消失させることができる.ヒトのジフテリア毒素受容体を発現しているマウスにジフテリア毒素を投与すると,筋線維芽細胞の数が減少した.これにより心筋梗塞による線維化が減少した102).
しかし,線維化が進行している心不全患者にCAR T細胞を投与することに対し懸念も提起されている103).CAR T細胞は筋線維芽細胞を攻撃するためのサイトカインを放出する.心不全患者の心臓は炎症が進行した状態にあることから,放出されたサイトカインは心不全の状態を悪化させるかもしれない.筋線維芽細胞の除去も問題になる可能性がある.筋線維芽細胞は,心臓の破裂を防ぐコラーゲンやフィブロネクチンなどのECMを産生している.したがって,筋線維芽細胞の除去を間違えると心臓の破裂を引き起こす.筋線維芽細胞の除去を行う時期が重要であり,その見分け方が難しい.さらなる問題点は,ヒトの筋線維芽細胞に特異的なマーカータンパク質が存在していないことである.特定の抗原は,CAR T細胞依存性治療の開発に必須なため,マーカータンパク質が利用できないと治療法自体が成り立たなくなる.これらの困難な点にもかかわらず,筋線維芽細胞の出現をみながら,CAR T細胞を用いて細胞数を適切に管理することで,副作用の少ない線維化の治療法になると期待されている.
CAR T細胞を用いた治療では,筋線維芽細胞に特異的な抗原および抗体を利用して細胞毒性を引き起こす作用を期待する.CAR T細胞を用いた治療法と似た方法として,二重認識タンパク質を利用した治療法が報告されている104).プログラム細胞死リガンド-1(PD-L1)とTGF-β(M7824)の二重阻害活性を持つタンパク質が,腫瘍の成長と転移の治療に用いられた.二重阻害剤は,TGF-β阻害剤を単独で使う治療よりも効果的であった.作用を局所にとどまらせるために,TGF-β阻害剤の作用特異性を高めることが期待できる.したがって,TGF-β阻害剤と筋線維芽細胞に特異的なタンパク質を認識する抗体を単一の分子に組み込むことが,線維化を治療するための新たなアプローチになる可能性がある.筋線維芽細胞に特異的な抗原の同定は,CAR T細胞を用いた治療のみならず,二重認識タンパク質によるTGF-β阻害作用を局所に制限することにつながり,それは線維化に対するTGF-β阻害剤の特異性を高めるのに役立つ.
6)レニン–アンジオテンシン–アルドステロン系に作用する薬剤
表2に抗線維化薬の臨床試験を示した.レニン–アンジオテンシン–アルドステロン系および他のシグナル伝達経路を標的とするさまざまな薬剤があり,線維化への効果が検討されている.なお,ここでは行われたすべての試験を網羅していない.また,表中の臨床試験は患者数が少ないことから,試験の結果に過度の期待を持つのは危険である.患者数が少ない場合,1人のばらつきが有意差に大きく寄与する.大規模な臨床試験での効果を検討する必要がある.
表2 臨床で抗線維化作用が検討された薬剤| 薬の作用点に基づく大まかな区分け | 使われた薬剤 | 処置の期間 | 患者数(n) | 主な知見 | 文献 |
|---|
| レニン–アンジオテンシン–アルドステロン系 | リシノプリル
ロサルタン
スピロノラクトン
エプレレノン | 6か月~24か月 | 19~153 | ・コラーゲン体積分率(CVF)の減少 | 107~114 |
| ・線維化の減弱 |
| ・血中のプロコラーゲンの一部配列が減少 |
| スタチン | アトルバスタチン | 6か月 | 56 | ・血中のプロコラーゲンの一部配列が減少 | 115 |
| ループ利尿薬 | トルセミド | 8か月 | 23~36 | ・コラーゲン体積分率(CVF)の減少 | 116, 117 |
| ・血中のプロコラーゲンの一部配列が減少 |
| ・左室の硬さの正常化 |
| cGMP選択的ホスホジエステラーゼ5A阻害剤 | シルデナフィル | 3か月 | 59 | ・TGF-βやMCP-1の減少 | 118, 119 |
| 文献120を改変. 略号:CVF:collagen volume fraction, MCP-1:monocyte chemoattractant protein-1. |
治療薬の最後に,抗線維化薬の開発がなぜ困難であるかについて,これまでの報告に基づいて紹介する.トラニラスト(アレルギー性疾患の治療に用いられ,TGF-β1の遊離抑制作用なども持つため線維芽細胞のコラーゲン合成を抑制する)やロスバスタチン(肝臓においてコレステロール合成経路の律速酵素であるHMG-CoA還元酵素を選択的・競合的に阻害し,コレステロール合成を抑制する)などのさまざまな薬剤は,動物モデルの線維化に効果を示す.しかしながら,これらの薬剤はヒトの線維化には効果を示さない.実際,線維化を治療するために開発された多くの薬剤が臨床試験では失敗している105).このことは,心臓の抗線維化薬の開発ではいくつかの乗り越えなくてはならない問題点があることを示している.第一に,動物モデルからヒトへの結果の外挿は適切ではない.ヒトでは,線維化はゆっくりと進行している.それは数年から数十年かかる.これに対し,動物モデルの線維化は数日から数か月で生じる.第二に,マウスとヒトの間には遺伝的な差異がある.人間の遺伝子の不均一性は,抗線維化作用を期待した薬物が効果を示さないことと関連している可能性がある.第三に,実験に使用した動物と線維化の患者の間には年齢の違いがある.モデルを作製する動物は通常若いのに対し,患者は多くの場合高齢である.さらに,多くの動物モデルにはさまざまな臨床上の設定が反映されていない.第四に,動物とヒトでは薬物代謝に違いがある.ヒトの線維化にはさまざまな因子が関わっており,動物モデルではこれらの因子を考慮していない場合がある.これらの問題点を乗り越え有益な結果を得るには,薬剤の種類や投与量,投与のタイミング,投与期間および患者の選択を含む臨床試験の注意深い設計が必要とされている.