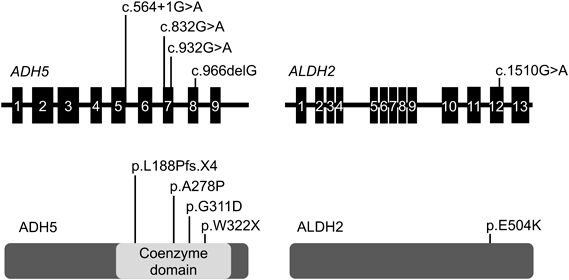
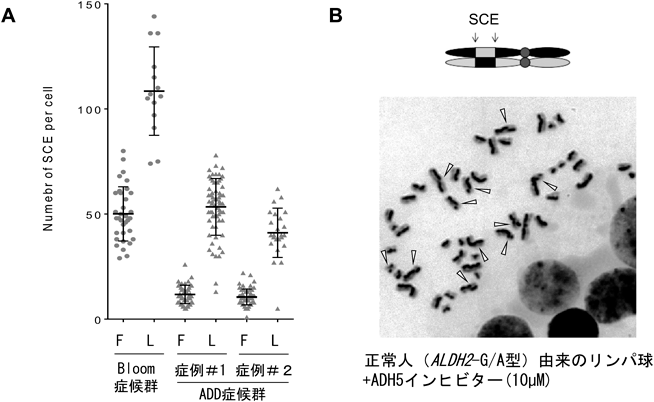
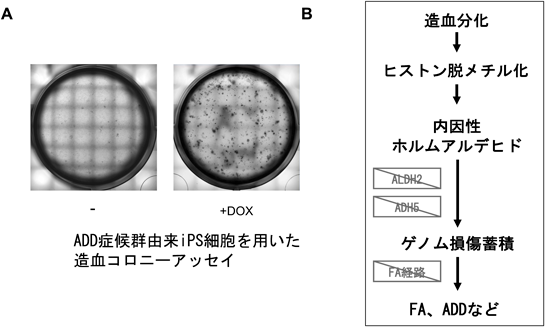
aldehyde degradation deficiency(ADD)症候群:アルデヒド代謝酵素ADH5/ALDH2欠損による新規遺伝性再生不良性貧血Aldehyde Degradation Deficiency (ADD) Syndrome: Impaired metabolism due to ADH5/ALDH2 mutations causes a novel inherited BMF syndrome
京都大学大学院生命科学研究科附属放射線生物研究センター晩発効果研究部門DNA損傷シグナル研究分野Laboratory of DNA Damage Signaling, Department of Late Effects Studies, Radiation Biology Center, Graduate School of Biostudies, Kyoto University ◇ 〒606–8501 京都市左京区吉田近衛町 ◇ Yoshidakonoecho, Sakyo-ku, Kyoto 606–8501, Japan
発行日:2022年2月25日Published: February 25, 2022