ミトコンドリア病とは,ミトコンドリアの機能障害によって引き起こされる多様な疾患群全体を指している.ミトコンドリアが独自に有するミトコンドリアDNA(mtDNA)にコードされた遺伝子は,すべてがミトコンドリアの内膜に埋め込まれた呼吸酵素複合体を構成するサブユニットか,それらを翻訳するために必須のtRNAまたはrRNAであるから,mtDNAへの病原性突然変異の蓄積はミトコンドリアのATP産生機能に影響を及ぼし,ミトコンドリア病の原因となる.しかし,ミトコンドリア病のすべてが,mtDNAの病原性突然変異によって引き起こされているわけではない.ミトコンドリアの機能はmtDNAだけでなく,核DNAによってコードされた1000以上の遺伝子によっても制御されていて,それらの突然変異もミトコンドリアの機能異常を引き起こしうるためだ.事実,現在までに,ミトコンドリアで機能する核DNAコードの遺伝子のうち少なくとも200程度について,その突然変異が実際にミトコンドリア病の原因になると報告されている.つまりミトコンドリア病には,mtDNAの病原性突然変異が原因となっているものと,核DNAの突然変異が原因となっているものの大きく分けて二つのタイプが存在する.本稿では,このうちmtDNAの突然変異によって引き起こされる病態と,その病態を理解する目的で作出された,突然変異型mtDNAを有するモデルマウスを用いた研究によって明らかになった知見について概説したい.
哺乳類のmtDNAは,16.5 kbほどの環状二本鎖構造である.ヒトの核ゲノムのサイズ(約3 Gb)と比較すると,わずか18万分の1にも満たない小さなゲノムだ.しかし,コードされている遺伝子のすべてが酸化的リン酸化に必須のものであり,Non-coding領域が少なく,ほぼ全長にわたって37個の遺伝子がすき間なく並んでいることから,mtDNAへの突然変異によるインパクトは,ゲノムサイズの小ささに反して大きいといえる.
mtDNAは,核DNAと比べると修復機構に乏しいといわれている.核DNAの場合には,ミスマッチ修復や塩基除去修復(変異箇所の塩基だけを除去し,正しい塩基に置換する),ヌクレオチド除去修復(変異箇所を含む複数の塩基をまとめて除去し,正しい塩基をつなぎ直す)など複数の修復機構が知られているが,mtDNAの修復はほとんどすべて塩基除去修復のみに依存している1).また,核DNAの複製は細胞分裂時にしか起こらないが,mtDNAの複製は細胞分裂時だけでなく間期にも一定の頻度で起きていることから,複製時のエラーも蓄積しやすい.こうした背景から,mtDNAは核DNAと比べると5~10倍程度,突然変異が入りやすいと考えられている.
一方で,こうしたmtDNAの“易変異性”を補完するかのような,mtDNA独自のシステムもある.それが,mtDNAの“多コピー性”である.細胞の種類にもよるが,mtDNAは1細胞あたり数百~数千コピー存在し,最も多い卵細胞では10万コピー前後に及ぶ.これはつまり,1000コピーあるうちのたとえば10コピー程度に仮に重大な突然変異が生じたとしても,残りの990コピーが正常であればミトコンドリアの機能にはまったく影響しないことを意味している.変異が容易に生じやすい分,コピー数を多くすることで変異による影響を希釈しているとも考えられ,優れた病態発症防止機構であるといえよう.すなわち,mtDNAの突然変異が原因となって現れている病態は,mtDNAの多コピー性による希釈効果を凌駕して病原性突然変異型mtDNAが蓄積した結果であると考えられる.
1)MELAS
ミトコンドリア病の中で最も患者数が多い疾患が,MELAS(mitochondrial myopathy, encephalopathy, lactic acidosis, and stroke-like episodes)2)である.大部分は小児期に発症し,全身の硬直やてんかん様発作,脳卒中様症状,反復性の頭痛や嘔吐などを主訴とする多臓器疾患で,多くは青年期までに運動機能や視力の低下,難聴などの神経症状を併発する.症状は次第に増悪し,壮年期を前に死亡することも少なくない3).ある調査では,MELAS患者の平均寿命は34.5±19歳(10.2~81.8歳の範囲)であったとしている4).
MELASと診断される80%程度の症例において,mtDNAのtRNALeu(UUR)遺伝子に突然変異が見つかるとされており,その大部分がA3243G変異である5).この変異の他に,同じtRNALeu(UUR)遺伝子上のT3271C変異やT3291C変異もMELASの原因として知られているが,A3243Gと比べると圧倒的に頻度は低い.tRNALeu(UUR)遺伝子以外の遺伝子の突然変異として,呼吸酵素複合体IのサブユニットであるND5をコードする遺伝子上のG13513A変異も,MELAS患者の10~15%程度で見つかる.この他,tRNAPheやtRNALysなどの変異もいくつかMELASの原因として報告されているが,いずれも症例は少ない.
これらの突然変異に共通しているのは,同一細胞内に野生型mtDNAと変異型mtDNAが共存したヘテロプラスミーとして検出される点である.つまり,患者ごとに,さらには同一患者内であっても組織を構成する細胞ごとに,変異率は異なっていて,それが疾患の重篤度を決める一つの要因になっている.細胞内のmtDNAすべてが変異型となるホモプラスミーとしての症例がないのは,これらの変異の病原性が強いためにホモプラスミーでは正常な細胞機能を維持できず,出生に至ることがないためであると考えられる.
2)MERRF
MELASに次いで患者数の多いミトコンドリア病が,MERRF(myoclonus epilepsy associated with ragged-red fibers)6)である.発症時期は小児~壮年期まで患者によってかなり幅があるが,多くは10歳前後で本疾患に特徴的な進行性のミオクローヌスてんかんがみられるようになる.ミオクローヌスとは,身体の一部の筋肉(手足や指,まぶたなど)が瞬間的にピクッと不随意に動く症状のことで,これが意識消失や全身のひきつけといったてんかん様発作を併発する.進行性なので,初めは疲れやすいといったさほど病的ではない症状だったものが,次第に筋力低下が著しくなり,歩行障害など日常生活にも影響が出るようになる.病名に含まれているragged-red fiber(RRF)は赤色ぼろ線維と訳される筋生検の病理所見で,ゴモリ・トリクローム染色という特殊染色を行うことによって観察される,筋組織中の異常ミトコンドリアの集積像である.赤色ぼろ線維はMERRF以外のミトコンドリア病,たとえば前述のMELASでも観察されるため,必ずしもMERRFだけに特異的な所見ではないが,ミトコンドリアの機能異常によって筋組織に大きな影響が出ていることを示す特徴的な指標であるといえる.
MELASの病因変異のほとんどがmtDNAのtRNALeu(UUR)遺伝子上に見いだされるが,MERRFの場合はtRNALys遺伝子の突然変異が原因であるケースが大部分を占める.代表的な突然変異はA8344G変異7, 8)で,MERRFと確定診断された症例の80%近くはこの変異が原因とされる.他に,同じtRNALys遺伝子上のT8356C変異9)やG8363A変異10)もMERRFの原因になるとされ,MERRF症例のほとんどでA8344G変異と併せたこれら三つの変異のいずれかが確認できる.
MELASの変異と同様に,MERRFの原因となるtRNALys遺伝子の突然変異も,やはりヘテロプラスミーとして存在している.
3)CPEO/KSS
三大病型の三つ目は,慢性進行性外眼筋麻痺と訳されるCPEO(chronic progressive external ophthalmoplegia)11)である.外眼筋麻痺は本来病名ではなく一つの症状を指すのだが,本疾患ではこの外眼筋麻痺と眼瞼下垂が主症状としてみられるために,症状名が病名として扱われている.これらの眼症状に加えて,心伝導障害や網膜色素変性を伴うCPEOの重症型を,KSS(Kearns-Sayre syndrome)12)と呼んで区別している.
CPEO患者の半数以上,そして重症型であるKSS患者のほとんどにおいて,mtDNAの欠失突然変異が見つかる.この欠失は数塩基程度の小さなものではなく,数千bpにも及ぶ大規模な欠失であり,ほとんどの症例において,欠失領域はmtDNAの8500番目前後の塩基~13500番目前後の塩基にかけての5000 bp前後を欠いている.多くの患者で共通した領域に欠失が認められるため,common deletionとも呼ばれる13).
ヒトmtDNAの全長が16.5 kb程度の大きさであることを考えると,その30%程度に相当する5000 bpの欠失は非常に大きい.この5000 bpの中には複数の構造遺伝子とtRNA遺伝子が含まれており,遺伝子が丸ごと複数欠落する状態になる.mtDNAにコードされた遺伝子はすべてミトコンドリアの正常な呼吸機能に必須であるため,欠失型mtDNAだけでは呼吸機能を維持できない.そのため,この欠失型mtDNAもすべての症例においてヘテロプラスミーで存在する.
1)Leber病
Leber病の正式な英語疾患名はLeber’s hereditary optic neuropathyで,LHONやLeber遺伝性視神経症,Leber視神経萎縮症などいくつか同義の呼称がある.10~30歳程度の若年成人期の男性に多く発症し,両眼の視力が亜急性に(数週間~数か月をかけて)低下して1年程度で視神経萎縮に至る.この視力障害に加えて,振戦や運動障害なども併発するケースも多い14).
Leber病の原因として,mtDNAのG11778A変異が最も多く報告されており,次いでT14484C変異,G3460A変異が見つかる.この三つの変異でLeber病患者全体の90%が説明できるが,アジア系民族の場合はとりわけG11778A変異の頻度が高く,日本人でもこの変異単独で90%を占めるとされる.MELASやMERRFの原因変異が特定のtRNA遺伝子に集中していたのと比べると,Leber病の原因変異はmtDNAの離れた場所に点在しているようにみえる.しかし,G11778Aは呼吸酵素複合体IのサブユニットであるND4遺伝子上,T14484C変異は同じ呼吸酵素複合体Iを構成する別のサブユニットであるND6遺伝子上,そしてG3460A変異もやはり呼吸酵素複合体IのサブユニットであるND1遺伝子上の変異であり,すべて呼吸酵素複合体Iを構成する構造遺伝子上の点突然変異であることがわかる.これらの変異の多くは,tRNA遺伝子の変異とは異なり細胞内のmtDNAすべてが変異型であるホモプラスミーとして見つかる.野生型mtDNAとのヘテロプラスミーで見つかる場合もあるが,そのような場合であっても変異型mtDNAの割合がかなり高い15).
2)Pearson病
Pearson病は鉄芽球性貧血,汎血球減少と膵外分泌不全を主症状とした,乳児期に発症する重篤なミトコンドリア病である.骨髄の塗抹標本に鉄染色を施して観察すると,ミトコンドリアに鉄が異常沈着して赤芽球(赤血球の前駆細胞)の核の周囲を取り囲んだ環状鉄芽球という特徴的な所見が認められることが多い.同時に,骨髄芽球(白血球の前駆細胞)の細胞質には通常はみられない空胞が多く観察される.膵臓で作られるリパーゼやキモトリプシンといった消化酵素が十分に分泌されず,消化不良による下痢が引き起こされる.この他,肝機能や腎機能も障害されて重篤な場合は死に至る.
Pearson病の原因は,CPEOやKSSと同じmtDNAの大規模欠失突然変異である.実際,Pearson病と診断されてから貧血症状が最も強く現れる乳幼児期を越えて生き残ったsurvivorの中には,その後KSSの病型へと移行していく例があると報告されており,Pearson病はCPEOやKSSのより若齢時に発症する重篤型と考えることもできる.
3)Leigh脳症
Leigh脳症は,以前は生前の診断が難しく,死後の脳の病理所見が診断の判断材料とされてきた.しかしMRIなどの画像診断技術が進歩したことによって,現在では大脳基底核や脳幹で特徴的な白質病変が鑑別できるようになり,生前でも診断が可能となってきた.この白質病変は,当該部位で神経細胞の壊死が起きていることを示しており,多くは乳幼児期に精神運動発達の遅延が認められ,知的退行(以前はできたことができなくなる)やけいれん発作,呼吸障害などの症状が発症する16).病態は発症時期が早いほど重篤であると考えられており,幼児期までに死に至ることも少なくない.
Leigh脳症はこれまでに紹介してきた他のミトコンドリア病と比較して,その原因となる遺伝子が多岐にわたることが知られている.最もよく知られているのは,ミトコンドリアの呼吸酵素複合体IVを組み立てる際に必須となる核DNAにコードされたタンパク質SURF1の突然変異であるが,その他にも呼吸酵素複合体のIやIIを形成する多くの核DNAコードのサブユニット遺伝子(NDUFS4, NDUFA2, NDUFS7, SDHAなど)の突然変異がLeigh脳症の原因になるとされている.mtDNAコードの遺伝子としては,呼吸酵素複合体VのサブユニットであるATP6遺伝子上のT8993G変異が最も多く報告されている.ただ,Leigh脳症の既知の原因遺伝子の約8割は核DNAコードのミトコンドリア関連遺伝子で,T8993GをはじめとするmtDNAの突然変異によるLeigh脳症は全体の2割程度であるとされている17).
ここまでに紹介した主なミトコンドリア病の各特徴をまとめた概略を,表1にまとめた.
表1 主なミトコンドリア病とその特徴| 疾患名 | ミトコンドリア病の「三大病型」 | Leber病 | Pearson病 | Leigh脳症 |
|---|
| MELAS | MERRF | CPEO/KSS |
|---|
| 主な症状 | 全身の硬直,てんかん様発作,脳卒中様症状,反復性の嘔吐や頭痛,運動機能低下,視力低下,難聴など | ミオクローヌスてんかん,進行性の筋力低下による歩行障害など.筋組織にRRFがみられる | 外眼筋麻痺,心伝導障害,網膜色素変性など | 亜急性の視力低下,視神経萎縮,振戦,運動障害等 | 鉄芽球性貧血,汎血球減少,膵外分泌不全,下痢,肝機能障害,腎機能障害など | 精神運動発達遅延,知的退行,けいれん発作,呼吸障害など |
| 主な原因 | mtDNAのtRNALeu (UUR) 遺伝子上のA3243G変異,T3271C変異,T3291C変異等 | mtDNAのtRNALys遺伝子上のA8344G変異,T8536C変異,G8363A変異等 | mtDNAのcommon deletion領域の大規模欠失突然変異 | mtDNAのG11778A変異,T14484C変異,G3460A変異など,呼吸酵素複合体Iのサブユニットをコードする構造遺伝子上の変異等 | mtDNAのcommon deletion領域の大規模欠失突然変異 | 多くは核DNAにコードされたミトコンドリア関連遺伝子の突然変異だが,mtDNAのATP6遺伝子上のT8993G変異等も原因となる |
| ヘテロ/ホモ | ヘテロプラスミー | ヘテロプラスミー | ヘテロプラスミー | ホモプラスミーが多い | ヘテロプラスミー | ヘテロプラスミー/ホモプラスミーいずれも報告あり |
ミトコンドリア病には,まだまだよくわかっていないことがたくさんある.
そもそも,正確な患者数の把握もままならない.なぜなら,ミトコンドリア病の症状が多様であり,診断自体が簡単ではないからだ.たとえばMELASの場合,てんかんのような症状を訴えて病院にかかったとしても,MELASによるてんかん様発作を,それ以外の要因によるてんかんと区別するのは難しく,本当はMELASが原因であるのに,一般的なてんかんと診断されている患者は一定数いると考えられる.ミトコンドリア病の確定診断には,血液のように採取しやすい検体を用いた検査に加えて,筋生検の採取など侵襲的な検査を必要とする場合もあり,ミトコンドリア病を強く疑わないとなかなか実施に踏み切れないという側面もある.ミトコンドリア病の診療経験が豊富な専門医は少なく,ミトコンドリア病という病気自体の認知度も高くない.そのため,本当はミトコンドリア病なのに,正しく診断されていない患者はかなりいるのではないかと想定される.ミトコンドリア病患者は人口5000人に1人いる程度と推計されている18)が,上記のような理由から,本当はもっと高い割合である可能性も大いにある.
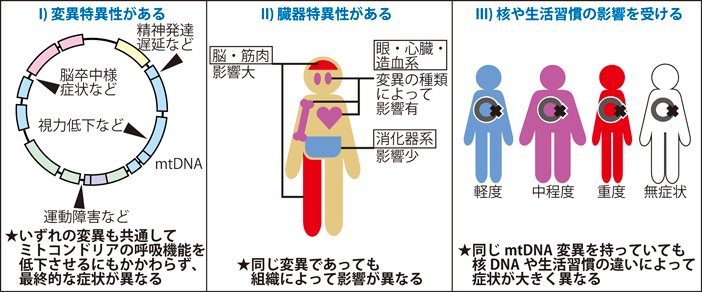
また,なぜ多様な病態が現れるのか,という点もミトコンドリア病をめぐる大きな謎の一つである.mtDNAにコードされた遺伝子は,すべてミトコンドリアの酸化的リン酸化に必須の遺伝子である.したがって,それらの遺伝子の機能を損なう病原性突然変異は,mtDNA上のどの遺伝子の突然変異であったとしても,ミトコンドリアのATP産生機能の低下という共通の一次現象を引き起こす.それなのに,結果として現れる病態には大きな多様性が生じる(図1-I).同じように変異を蓄積していても,病態が現れやすい臓器とそうでない臓器があり,変異の種類ごとに症状の現れやすさには一定の臓器特異性がある(図1-II).さらに,同じ突然変異を有する患者どうしであっても,患者ごとに病態が現れる臓器や,観察される病態の質や程度が大きく異なることもあり,患者ごとの核DNAの遺伝的背景や生活習慣などによる影響を受けている可能性が考えられる(図1-III).
その一方で,これまで紹介してきたとおり,変異の種類ごとに現れる病型に一定の型があることも事実である.たとえば,tRNALeu(UUR)遺伝子の突然変異はMELASの特徴的な病態を引き起こしやすく,tRNALys遺伝子の突然変異はミオクローヌスてんかんを主としたMERRF病態の原因とされる.common deletion領域の大規模欠失突然変異の病態は眼に現れやすい.なぜ,どの突然変異もATP産生の低下を引き起こすという点は共通しているのに,病態には変異の種類ごとにある程度の決まった型があり,変異によって病態が現れやすい臓器とそうでない臓器があるのだろうか.そして,同じtRNA遺伝子の突然変異なのに,tRNALeu(UUR)遺伝子とtRNALys遺伝子が異なる病態の原因となっている理由も定かでない.どのtRNAであっても,機能不全に陥ればミトコンドリア内のタンパク質合成に大きく影響を及ぼすはずであるから,単純にミトコンドリア内のタンパク質合成能が低下すること「だけ」がMELASやMERRFの臨床症状を引き起こしているわけではなさそうだ.tRNALeu(UUR)遺伝子やtRNALys遺伝子の突然変異に特異的な,何らかの病態発症機構があると考えられるが,その詳細はほとんど明らかにされていない.
6. ミトコンドリア病モデルマウスの必要性とその樹立における壁
こうした数多くのミトコンドリア病をめぐる謎を解明するには,臨床の研究や調査だけでは限界がある.冒頭でも述べたとおり,ミトコンドリアの機能はmtDNAにコードされた遺伝子だけではなく,核DNAにコードされた1000以上の遺伝子によっても支えられているため,核背景によっては,特定のmtDNAの突然変異による影響を受けやすい場合とそうでない場合があるはずだ.このことは,同じ変異を持っていながら,病態の質や程度が異なる患者がいるという事実に対する一つの説明になると考えられているが,ヒトは個人ごとに核背景が異なるため,それを実際に検証するのは難しい.そのため,核背景が均一なモデル動物を用いた検証が必須となる.
また,有害なmtDNA変異の多くは,野生型mtDNAと細胞内で共存するヘテロプラスミー状態で存在している.それはすなわち,臓器ごと,細胞ごとに変異率が異なることを意味している.ミトコンドリア病では,ATP要求性の高い神経や筋で症状が現れやすいため,mtDNA変異と病態との関係性を精査する上で,実際に症状が出ている組織での変異型mtDNAの割合やミトコンドリアの機能の低下度合いなどは重要な情報である.しかし,ヒト患者に対して視神経が侵されているからといってその神経を採取したり,心不全がみられたからといって心筋を取ったりするわけにはいかない.モデル動物であれば,病態が現れる前後での疾患組織の採取や検査も可能であるから,より直接的に病態発症機構に迫ることができる.
こうした背景から,ミトコンドリア病の病態発症機構の解明のために,mtDNAに病原性突然変異をもち,それによって実際にヒトと類似した病態を示すミトコンドリア病モデルマウスの樹立が強く求められてきた.ゲノム編集技術が大幅に進歩した現代において,そうしたモデルマウスの樹立は一見難しくないように思える.ところが,ミトコンドリア病モデルマウスの樹立例は世界的にみてもいまだかなり限られているのが実情である.
その理由は,ひとえにmtDNAの人為的改変の難しさにある.核膜に包まれているとはいえ,かなり自由に物質を通す核膜孔があり,細胞分裂時には核膜ごと消失することによって容易にベクターがアクセス可能な核DNAとは異なり,mtDNAは細胞周期を通じて完全な二重膜に覆われている.ミトコンドリアで機能するタンパク質輸送のためのミトコンドリア移行シグナルを付加すれば,ある程度の大きさのタンパク質をミトコンドリア内に輸送させることは可能となりつつある19)ものの,それでもまだその自由度はかなり低い.さらに,mtDNAの多コピー性が大きな障壁となる.核DNAにコードされた遺伝子の場合,基本的に同一遺伝子は父方由来と母方由来の2コピーであるため,片側のアリルに変異を挿入したりノックアウトしたりすれば,ヘテロの変異体が得られる.仮にこのヘテロ変異体が1個体しか得られなかったとしても,これを交配させることで次世代では半数の個体がヘテロ変異体となり,このヘテロ変異体どうしの交配によって孫の代ではホモの変異体を得ることが期待できる.ところがmtDNAの場合は,1細胞で数百~数千コピーも同じ遺伝子が存在していることになるため,仮に何らかのベクター的なものをミトコンドリア内に輸送させることができたとしても,数コピーのmtDNAを改変した程度では到底細胞の機能に影響するほどの変異率を達成できない.次世代にも変異型mtDNAを伝達させようと考えたら,未受精卵あるいは初期胚のmtDNAを改変させる必要があるわけだが,卵は一般的な体細胞と比べて段違いにmtDNAのコピー数が多いため,非常に高効率の変異挿入が要求される.ごく最近になって,mtDNAの編集を可能にする新しい技術がいくつか報告されてきてはいる20–22)ものの,mtDNAの人為的な改変はいまだにかなり困難であり,ミトコンドリア病モデルマウスの樹立もなかなか思うように進んでいない.
しかし,こうした困難を乗り越えて樹立されたモデルマウスの解析によって,ミトコンドリア病を理解する上で重要な機構が明らかになってきた.
7. ミトコンドリア病モデルマウスとそこから明らかになった機構
1)mito-miceΔ:世界初のミトコンドリア病モデルマウス
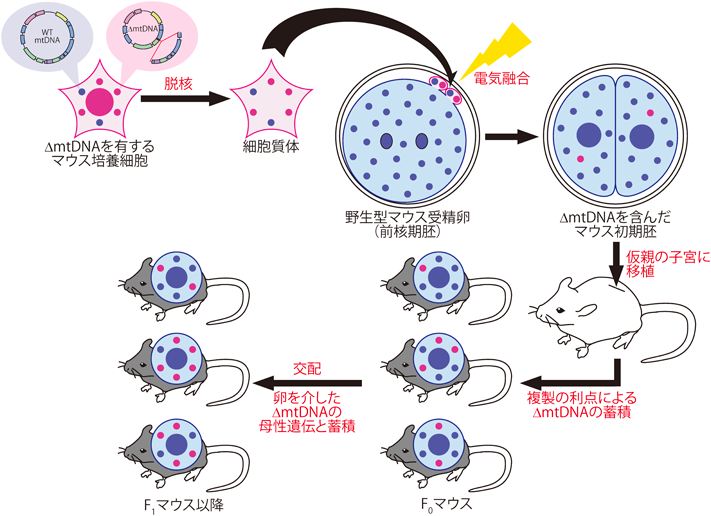
mito-miceΔは,CPEOやKSS, Pearson病の原因とされるヒトmtDNAのcommon deletion領域とほぼ同じ領域を欠失した,マウスの大規模欠失突然変異型mtDNA(ΔmtDNA)を有するマウスである23).2000年に報告されたこのマウスは,mtDNAの突然変異を原因としてミトコンドリア病患者で観察されるのと類似した病態を呈する,初のミトコンドリア病モデルマウスである.
本マウスは,人為的にmtDNAを改変したのではなく,マウス培養細胞中に偶然見つかったわずかなΔmtDNAをマウス個体に導入して作出された(図2).CPEOやKSSなどの患者と同じようにΔmtDNAをヘテロプラスミーで有しており,個体ごとはもちろんのこと,同一個体内でも組織や細胞ごとに,ΔmtDNAの含有率が異なっている.
mito-miceΔの主要な病態は心不全および腎不全である23)が,その他にもΔmtDNAの含有率が高い個体では妊孕性の低下24)や貧血25),長期記憶の低下26),低体重,低血糖,高乳酸血症27)といった多様な病態が現れる.
mtDNAに大規模欠失突然変異を有する点,変異がヘテロプラスミーで存在する点,変異率が高いと心不全が現れる点など,本マウスはヒトのKSSと類似した性質を有している.一方で,外眼筋麻痺や眼瞼下垂といったCPEOに特徴的な眼科症状はみられず,ヒトではさほど報告例の多くない腎不全が主症状として現れるなど,CPEO/KSSとは異なる側面もある.
2)ミトコンドリア間相互作用
mito-miceΔの病態解析を通じて,ミトコンドリアには優れた病態発症抑制機構があることがわかってきた.
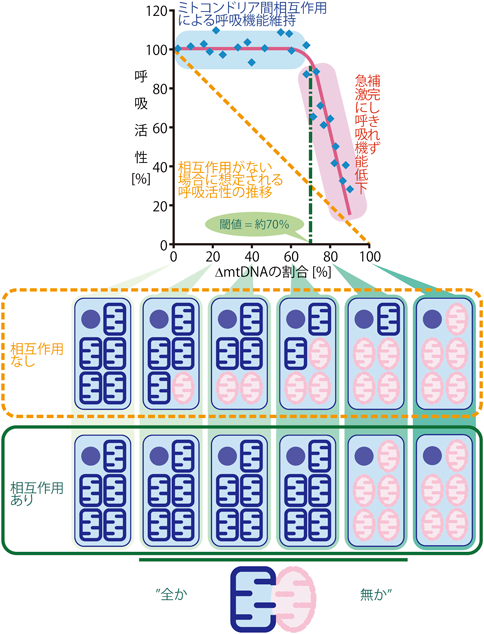
ヘテロプラスミーの病原性突然変異の場合,当然変異率が高い方が,低い場合よりも病態は強くなると考えられる.普通に考えれば,ミトコンドリアの呼吸機能は,mtDNAの変異率の増加に伴って直線的に低下するのではないかと予想される(図3,破線のグラフ).しかし,ΔmtDNAを有する細胞の呼吸機能を実際に調べてみると,ΔmtDNAの含有率が70%程度までは呼吸機能はほとんど野生型と同等であり,変異率が70%を超えたあたりから急速に呼吸機能が低下するということがわかった(図3,実線のグラフ).mito-miceΔの心筋を電子顕微鏡で観察すると,ある細胞には見かけ上正常なミトコンドリア「だけ」が存在し,隣り合う別の細胞には膜構造の形態が変化し,呼吸機能も低下した異常なミトコンドリア「だけ」が存在していて,正常と異常が同一細胞内で混在することはないという,意外な観察結果が得られた28).見かけ上正常なミトコンドリアしかないからといって,その細胞にΔmtDNAがないわけではない.逆に,異常なミトコンドリアしかないからといって,その細胞のmtDNAがすべてΔmtDNAになったわけでもない.つまり,mtDNA分子としてはヘテロなのだが,ミトコンドリアとしてはホモ(「正常か異常か」の「全か無か」状態)な状態になっているのである(図3下部,実線で囲んだ「相互作用あり」の部分参照).これは,ΔmtDNAの割合がある程度蓄積するまでは,ミトコンドリアが細胞内で融合・分裂を繰り返すことによって内容物を均質化し,不足を補いあって細胞全体としては正常なミトコンドリア機能を保っているが,ΔmtDNAの含有率が高くなりすぎると,限られた資源を細胞内のミトコンドリアが奪い合う形になって一気に全ミトコンドリアが機能不全に陥る,という「ミトコンドリア間相互作用」という仕組みの存在を強く示唆している.これによって,ある閾値(ΔmtDNAの場合は70%前後)までは細胞としては正常な呼吸機能が維持され,閾値を超えると一気に呼吸機能が低下するという実験結果も合理的に説明される.
「2. mtDNAとその突然変異」のところでもふれたとおり,mtDNAは多コピーなので,1000コピー中10や20コピーが病原性突然変異を有していても大きな影響はないだろう,ということは容易に想像できる.しかし実際には,この相互作用があるおかげで,1000コピー中実に700コピーが変異型であっても細胞機能が維持されるというのは,驚きに値する.
この相互作用はマウスだけでなくヒトでも機能していると考えられており,ミトコンドリア病の複雑な病態発症機構の一端を説明するものだと考えられている.
3)核–ミトコンドリア間クロストーク
mito-miceΔは一般的な野生型マウスの系統としてよく用いられるC57BL/6系統の核背景で維持されている.この核背景を別の疾患を発症する核背景に置き換えたり,核にコードされた特定のミトコンドリア関連遺伝子を機能不全にしたり,核背景を変えなくても飼育条件を変えたりすると,通常のmito-miceΔでは観察されなかった病態が現れることがあるということが,最近次第に明らかになってきた(未発表).これらの結果は,変異型mtDNAの種類は同じであっても,核背景や飼育環境を変えることによって異なる病態が現れることを示している.ヒトに置き換えれば,特定の遺伝的背景あるいは生活習慣などによって,変異型mtDNAの影響が大きく異なる可能性があるという,図1-IIIで予測された機構が実際に存在することを強く示唆している.
こうした核–ミトコンドリア間クロストークはミトコンドリア病の病態理解に欠かせない概念であると思われるが,ヒトの患者では解析が困難な要素でもあることから,モデルマウスを用いた研究による理解の進展が強く望まれる.
ミトコンドリアは酸化的リン酸化によってATP産生を担うという,高等動物のあらゆる細胞にとって必須の役割を担っていることから,その機能異常による影響は非常に大きい.
なぜその変異によって,なぜその組織で,なぜその病態が現れるのか,その病態発症機構には未知の部分が多く,有効な治療法の確立のためにも,臨床における患者研究と同時に,モデルマウス等を活用した基礎研究のますますの進展が期待される.
引用文献References
1) Stumpf, J.D. & Copeland, W.C. (2011) Mitochondrial DNA replication and disease: Insights from DNA polymerase γ mutations. Cell. Mol. Life Sci., 68, 219–233.
2) Pavlakis, S.G., Phillips, P.C., DiMauro, S., De Vivo, D.C., & Rowland, L.P. (1984) Mitochondrial myopathy, encephalopathy, lactic acidosis, and strokelike episodes: A distinctive clinical syndrome. Ann. Neurol., 16, 481–488.
3) Yatsuga, S., Povalko, N., Nishioka, J., Katayama, K., Kakimoto, N., Matsuishi, T., Kakuma, T., & Koga, Y. (2012) MELAS: A nationwide prospective cohort study of 96 patients in Japan. Biochim. Biophys. Acta, 1820, 619–624.
4) Kaufmann, P., Engelstad, K., Wei, Y., Kulikova, R., Oskoui, M., Sproule, D.M., Battista, V., Koenigsberger, D.Y., Pascual, J.M., Shanske, S., et al. (2011) Natural history of MELAS associated with mitochondrial DNA m.3243A>G genotype. Neurology, 77, 1965–1971.
5) Goto, Y., Nonaka, I., & Horai, S. (1990) A mutation in the tRNA(Leu)(UUR) gene associated with the MELAS subgroup of mitochondrial encephalomyopathies. Nature, 348, 651–653.
6) Velez-Bartolomei, F., Lee, C., & Enns, G.(2021) MERRF in GeneReviews (Adam, M. P., et al. eds.), University of Washington, Seattle, WA.
7) Shoffner, J.M., Lott, M.T., Lezza, A.M., Seibel, P., Ballinger, S.W., & Wallace, D.C. (1990) Myoclonic epilepsy and ragged-red fiber disease (MERRF) is associated with a mitochondrial DNA tRNA(Lys) mutation. Cell, 61, 931–937.
8) Yoneda, M., Tanno, Y., Horai, S., Ozawa, T., Miyatake, T., & Tsuji, S. (1990) A common mitochondrial DNA mutation in the t-RNA(Lys) of patients with myoclonus epilepsy associated with ragged-red fibers. Biochem. Int., 21, 789–796.
9) Zeviani, M., Muntoni, F., Savarese, N., Serra, G., Tiranti, V., Carrara, F., Mariotti, C., & DiDonato, S. (1993) A MERRF/MELAS overlap syndrome associated with a new point mutation in the mitochondrial DNA tRNA(Lys) gene. Eur. J. Hum. Genet., 1, 80–87.
10) Ozawa, M., Nishino, I., Horai, S., Nonaka, I., & Goto, Y.I. (1997) Myoclonus epilepsy associated with ragged-red fibers: A G-to-A mutation at nucleotide pair 8363 in mitochondrial tRNA(Lys) in two families. Muscle Nerve, 20, 271–278.
11) Hansen, A.C. & Logan, J.H. (1966) Chronic progressive external ophthalmoplegia. A review and case report. J. Natl. Med. Assoc., 58, 436–441.
12) Butler, I.J. & Gadoth, N. (1976) Kearns-Sayre syndrome. A review of a multisystem disorder of children and young adults. Arch. Intern. Med., 136, 1290–1293.
13) Schon, E.A., Rizzuto, R., Moraes, C.T., Nakase, H., Zeviani, M., & DiMauro, S. (1989) A direct repeat is a hotspot for large-scale deletion of human mitochondrial DNA. Science, 244, 346–349.
14) Blum, M., Hykin, P.G., Sanders, M., & Völcker, H.E. (1992) Theodor Leber: A founder of ophthalmic research. Surv. Ophthalmol., 37, 63–68.
15) Jha, R.K., Dawar, C., Hasan, Q., Pujar, A., Gupta, G., Vishnu, V.Y., Kekunnaya, R., & Thangaraj, K. (2021) Mitochondrial genetic heterogeneity in leber’s hereditary optic neuropathy: Original study with meta-analysis. Genes (Basel), 12, 1300.
16) Leigh, D. (1951) Subacute necrotizing encephalomyelopathy in an infant. J. Neurol. Neurosurg. Psychiatry, 14, 216–221.
17) Baertling, F., Rodenburg, R.J., Schaper, J., Smeitink, J.A., Koopman, W.J., Mayatepek, E., Morava, E., & Distelmaier, F. (2014) A guide to diagnosis and treatment of Leigh syndrome. J. Neurol. Neurosurg. Psychiatry, 85, 257–265.
18) Thorburn, D.R. (2004) Mitochondrial disorders: Prevalence, myths and advances. J. Inherit. Metab. Dis., 27, 349–362.
19) Bacman, S.R., Williams, S.L., Pinto, M., & Moraes, C.T. (2014) The use of mitochondria-targeted endonucleases to manipulate mtDNA. Methods Enzymol., 547, 373–397.
20) Mok, B.Y., de Moraes, M.H., Zeng, J., Bosch, D.E., Kotrys, A.V., Raguram, A., Hsu, F., Radey, M.C., Peterson, S.B., Mootha, V.K., et al. (2020) A bacterial cytidine deaminase toxin enables CRISPR-free mitochondrial base editing. Nature, 583, 631–637.
21) Hidaka, T., Hashiya, K., Bando, T., Pandian, G.N., & Sugiyama, H. (2021) Targeted elimination of mutated mitochondrial DNA by a multi-functional conjugate capable of sequence-specific adenine alkylation. Cell Chem. Biol.
22) Zekonyte, U., Bacman, S.R., Smith, J., Shoop, W., Pereira, C.V., Tomberlin, G., Stewart, J., Jantz, D., & Moraes, C.T. (2021) Mitochondrial targeted meganuclease as a platform to eliminate mutant mtDNA in vivo. Nat. Commun., 12, 3210.
23) Inoue, K., Nakada, K., Ogura, A., Isobe, K., Goto, Y., Nonaka, I., & Hayashi, J.I. (2000) Generation of mice with mitochondrial dysfunction by introducing mouse mtDNA carrying a deletion into zygotes. Nat. Genet., 26, 176–181.
24) Nakada, K., Sato, A., Yoshida, K., Morita, T., Tanaka, H., Inoue, S., Yonekawa, H., & Hayashi, J. (2006) Mitochondria-related male infertility. Proc. Natl. Acad. Sci. USA, 103, 15148–15153.
25) Inoue, S., Yokota, M., Nakada, K., Miyoshi, H., & Hayashi, J. (2007) Pathogenic mitochondrial DNA-induced respiration defects in hematopoietic cells result in anemia by suppressing erythroid differentiation. FEBS Lett., 581, 1910–1916.
26) Tanaka, D., Nakada, K., Takao, K., Ogasawara, E., Kasahara, A., Sato, A., Yonekawa, H., Miyakawa, T., & Hayashi, J. (2008) Normal mitochondrial respiratory function is essential for spatial remote memory in mice. Mol. Brain, 1, 21.
27) Ogasawara, E., Nakada, K., & Hayashi, J. (2010) Lactic acidemia in the pathogenesis of mice carrying mitochondrial DNA with a deletion. Hum. Mol. Genet., 19, 3179–3189.
28) Nakada, K., Inoue, K., Ono, T., Isobe, K., Ogura, A., Goto, Y.I., Nonaka, I., & Hayashi, J.I. (2001) Inter-mitochondrial complementation: Mitochondria-specific system preventing mice from expression of disease phenotypes by mutant mtDNA. Nat. Med., 7, 934–940.
著者紹介Author Profile
 石川 香(いしかわ かおり)
石川 香(いしかわ かおり)筑波大学生命環境系助教.博士(理学).
略歴2009年3月に筑波大学大学院生命環境科学研究科情報生物科学専攻を修了,博士(理学)取得.09年4月~14年3月まで,武田薬品工業株式会社医薬研究本部に勤務し,14年4月より現職.
研究テーマと抱負ミトコンドリアDNAの突然変異やミトコンドリアの機能変化が細胞や生体の機能に及ぼす影響を,モデルマウスや培養細胞を用いて研究しています.
ウェブサイトhttps://www.biol.tsukuba.ac.jp/nakada_ishikawa/index.html
趣味ゲーム(ドラゴンクエスト(DQ)),音楽鑑賞(主にDQの曲).No DQ Music, No Life!