細胞内のタンパク質は主に二つの経路(ユビキチン・プロテアソーム系とオートファジー・リソソーム系)で分解される.ユビキチン・プロテアソーム系はポリユビキチン鎖修飾を受けたタンパク質をプロテアソームで分解する経路であり,タンパク質を個別に分解する機構である18–21).もう一つのオートファジー・リソソーム系は,隔離膜によって取り囲んだ細胞小器官などをリソソームで一括して分解する機構である22, 23).本稿で紹介するタンパク質分解技術は,これら細胞にもともと備わっているタンパク質分解機構を利用して,標的とするタンパク質を分解する技術である.ユビキチン・プロテアソーム系を利用して標的タンパク質を分解する化合物の開発が先行して進んできたが,最近ではオートファジー・リソソーム系を利用してタンパク質分解を誘導する化合物の報告も増えつつある.
標的タンパク質を選択的に分解する化合物は,そのまま医薬品のリード化合物となる可能性もあるが,基礎研究のツールとしても有用である.ゲノム編集またはRNA干渉法で遺伝子発現を抑制することによって現れる表現型の変化を解析することは,目的とするタンパク質の機能解析によく用いられる常套手段である.しかしこれらタンパク質の生合成をブロックする方法では,標的タンパク質の発現量が変化するまでに比較的長い時間(数日)を必要とするため,その間に細胞にさまざまな二次的変化が生じてしまうことも多い.これに対して化合物による標的タンパク質の分解は数分から数時間で起こるため,タンパク質の消失(減少)による変化をより直接的に解析することができる.オーキシンデグロン法24, 25),dTAG(degradation tag)法26)などが知られているが,これらの技術の詳細については他の文献を参照していただきたい.
タンパク質分解を誘導する化合物はその構造と作用機序によっていくつかのカテゴリーに分類される.ここではその分類ごとに代表的な化合物と分解機構などについて述べる.
1)E3モジュレーター(分子糊)
E3モジュレーターはE3ユビキチンリガーゼ(以下E3)に結合して,その基質特異性を変化させる化合物である.植物ホルモンのオーキシンはCRLユビキチンリガーゼ複合体の基質認識サブユニットTIR1に結合すると,AUX/IAAタンパク質のユビキチン化と分解を引き起こす27, 28).AUX/IAAはオーキシン応答遺伝子の発現を抑制しているため,AUX/IAAを分解することによってオーキシン応答遺伝子の発現が誘導される.
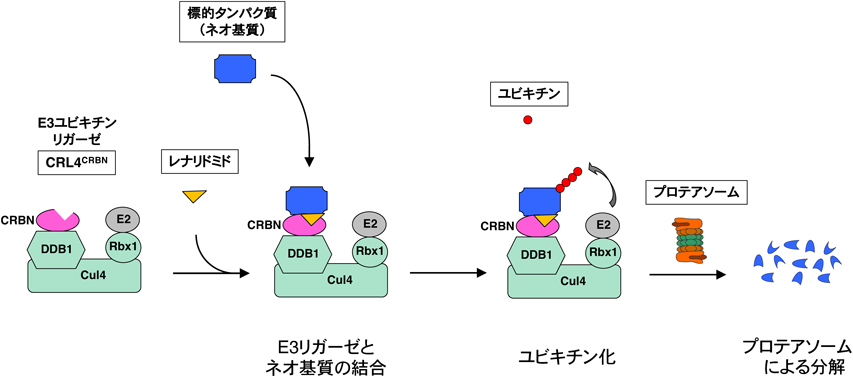
ヒトの医薬品として開発された化合物では,レナリドミドなどのサリドマイド類が同様な作用機序で標的タンパク質の分解を引き起こす(図1).サリドマイドは過去に催奇性が問題となったが,免疫調節機能などの有用な薬理作用が再び注目され,厳格な安全管理の下で多発性骨髄腫などの治療に使用されるようになった.その後サリドマイドの直接の標的分子がユビキチンリガーゼCRBNであり29),レナリドミド,ポマリドミドなどの類縁化合物も含めてE3モジュレーター作用によりイカロスファミリー転写因子(IKZF1, IKZF3)などのタンパク質を分解することが明らかになった30–32).化合物の側鎖を修飾することにより分解するタンパク質(ネオ基質)を変化させることができるが33, 34),現在の技術ではネオ基質を予測することはできない.
レナリドミドと同様にE3モジュレーター活性を示す化合物として,インディスラムなどのスルホンアミド化合物が知られている.インディスラムはDCAF15に結合し,スプライシング制御因子RBM39/CAPERαの分解を誘導する35, 36).
これらの化合物は,E3とネオ基質タンパク質を結合させる糊のような働きをすることから,分子糊(molecular glue)と呼ばれることもある.上記三つとは作用点が異なるが,CDK阻害剤CR8はユビキチンリガーゼのアダプタータンパク質DDB1とCDK12を結合させる分子糊として働き,CDK12と結合しているサイクリンKのユビキチン化と分解を誘導することが報告されている37).またBCL6阻害剤BI-3802は,BCL6タンパク質の重合を仲介する分子糊として作用し,SIAH1によるユビキチン化とプロテアソームによる分解を誘導する38).
2)疎水性タグ化合物
エストロゲン受容体を分解するフルベストラントはβエストラジオールに疎水性の側鎖が付加した構造をしており,エストロゲン受容体に結合すると受容体の構造変化を引き起こして分解を誘導する39).乳がんの治療薬として上市されているが,フルベストラントは分解を意図して開発された化合物ではなく,後にエストロゲン受容体を分解することが明らかになった40).同様なカテゴリーの化合物としてBoc3Arg,アダマンタンなどの疎水性残基を利用した化合物が報告されている41–44).化合物の結合によって構造変化を起こしたタンパク質が細胞内のタンパク質品質管理機構によって認識され分解に至ると考えられているが,分解に関与するユビキチンリガーゼなどの詳しいメカニズムはわかっていない.この方法で分解できる標的タンパク質は今のところエストロゲン受容体,アンドロゲン受容体などの一部のタンパク質に限られているが,Haloタグなどを利用して化合物を標的タンパク質に共有結合させると多くのタンパク質を分解することができる.
3)IAPアンタゴニスト
IAP(inhibitor of apoptosis protein)は細胞死を阻害し,炎症の制御に重要な役割を果たす一群のタンパク質であるが45, 46),そのうちcIAP1, cIAP2, XIAPなどのファミリーメンバーはE3活性を示す.IAPアンタゴニストはこれらIAPのBIR3ドメインに結合してその機能を阻害するだけでなく,cIAP1などのユビキチン化と分解を引き起こす.一部のがん細胞はIAPの過剰発現により細胞死を免れているため,IAPアンタゴニストは新しい作用機序を持つ抗がん剤として期待され,多くのIAPアンタゴニストが開発されてきた45, 47–52).しかしIAPアンタゴニストが分解を誘導できるのはcIAP1, cIAP2などの一部のIAPに限られている.
4)キメラ化合物(PROTAC, SNIPER)
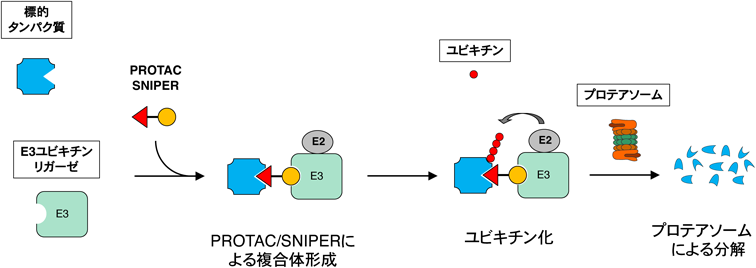
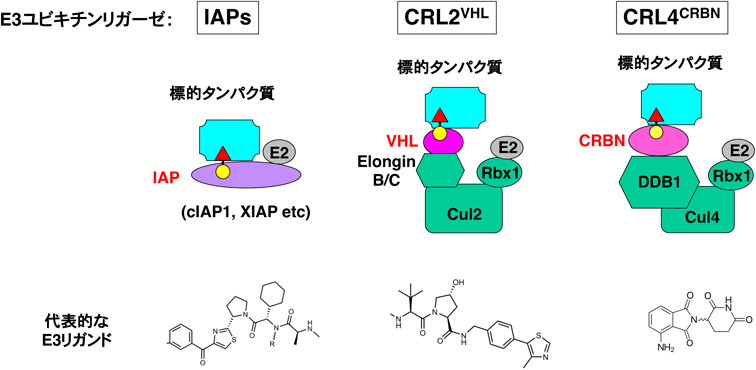
目的とする標的タンパク質の分解に現在最もよく利用されているのが,PROTAC, SNIPERなどのキメラ化合物である.最近はこれらのキメラ化合物をまとめてPROTACと総称することが多いが,本稿ではPROTACの中でIAPを利用して分解を誘導する化合物をSNIPERとして扱う13, 14).PROTAC/SNIPERはE3に結合するリガンドと標的タンパク質に結合するリガンドをつないだキメラ構造をしており,細胞内で標的タンパク質とE3を近接させることにより標的タンパク質のユビキチン化とプロテアソームによる分解を誘導する(図2).標的リガンドを取り替えることによって任意のタンパク質を分解するキメラ化合物を合理的に設計できるという特徴がある.初期のPROTACはE3リガンドにリン酸化ペプチドなどを利用していたため無細胞系で活性を示すにとどまっていたが53),両方のリガンドに小分子化合物を導入したPROTAC/SNIPERが開発され54–56),培養細胞系で標的タンパク質の分解活性を示すようになった.その後E3リガンドの改良によりin vivoでも標的タンパク質の分解活性と薬理活性を示すPROTAC/SNIPERが開発され57–62),創薬の新しいプラットフォーム技術として注目されるようになった63).現在のところ,標的タンパク質の分解に利用されるE3はCRL4CRBN,CRL2VHL,IAPが多いが,他のE3を利用して分解する化合物の報告も増えてきている55, 64–69).
5)脱ユビキチン化酵素阻害剤
ユビキチン化の逆反応を阻害することによって標的タンパク質の分解を誘導する化合物も報告されている.細胞内のユビキチン化反応は可逆的な反応であり,標的タンパク質に形成されたポリユビキチン鎖は脱ユビキチン化酵素によって除去される70, 71).細胞内の多くのタンパク質はユビキチン化が引き金となって分解されるが,一部のタンパク質は常にユビキチン化を受けていながら脱ユビキチン化酵素の作用によって分解を免れている.細胞には約100種類の脱ユビキチン化酵素があるが,たとえば慢性骨髄性白血病の融合タンパク質BCR-ABLはUSP25による脱ユビキチン化によって細胞内で分解を免れているため,USP25阻害剤はBCR-ABLの分解を誘導する72).また別の脱ユビキチン化酵素USP19の発現を抑制するとユーイング肉腫の融合タンパク質EWS-FLI1が減少し,肉腫の増殖が抑制される73).その他にもUSP9Xを阻害するWP1130はERGタンパク質の分解を引き起こすことが報告されている74).脱ユビキチン化酵素阻害剤は多くのタンパク質の分解に適用できるわけではないが,がん細胞の増殖に重要な変異タンパク質のいくつかは脱ユビキチン化酵素を阻害することによって分解できると考えられる.
6)オートファジー・リソソーム系による分解を誘導する化合物
上記の化合物はいずれもユビキチン・プロテアソーム系に作用して標的タンパク質の分解を誘導するが,オートファジー・リソソーム系を利用してタンパク質分解を誘導する化合物も報告されている.
細胞内に侵入したA群連鎖球菌はオートファジーによって分解されるが,その際に菌表面がS-グアニル化修飾されこれを目印として隔離膜が形成される.AUTAC(autophagy-targeting chimera)は,S-グアニル基を模したp-fluorobenzylguanineと標的リガンドをリンカーでつないだキメラ化合物で,標的タンパク質やミトコンドリアの周りに隔離膜形成を誘導しオートファジーで分解する75, 76).
マンノース6-リン酸受容体などの細胞膜受容体は刺激によって細胞内に取り込まれリソソームで分解される.LYTAC(lysosome-targeting chimera)は,マンノース6-リン酸受容体のリガンド(ポリペプチドにマンノース6-リン酸を数十個結合した分子)に抗体を結合したキメラ分子で,抗体が結合するタンパク質(細胞外もしくは細胞膜上のタンパク質)をマンノース6-リン酸受容体とともに細胞内に取り込んでリソソームで分解する77).
またATTEC(autophagosome-tethering compound)は,伸長したポリグルタミンとLC3の両者に結合活性を示す化合物で,分子糊のように作用して変異型タウなどの凝集性タンパク質を分解する活性を示すことが報告されている78, 79).
以上のようにタンパク質分解を誘導するさまざまな化合物が開発されてきたが,目的のタンパク質を分解する化合物を合理的に設計可能なPROTAC/SNIPERをベースにした創薬研究が現在活発に行われている.時間的には少し遡るが,以下の項では我々の研究室で行ってきたSNIPER開発の経緯とPROTAC/SNIPERの特徴などについて述べる.
1)MeBSによるcIAP1分解からSNIPER化合物の着想
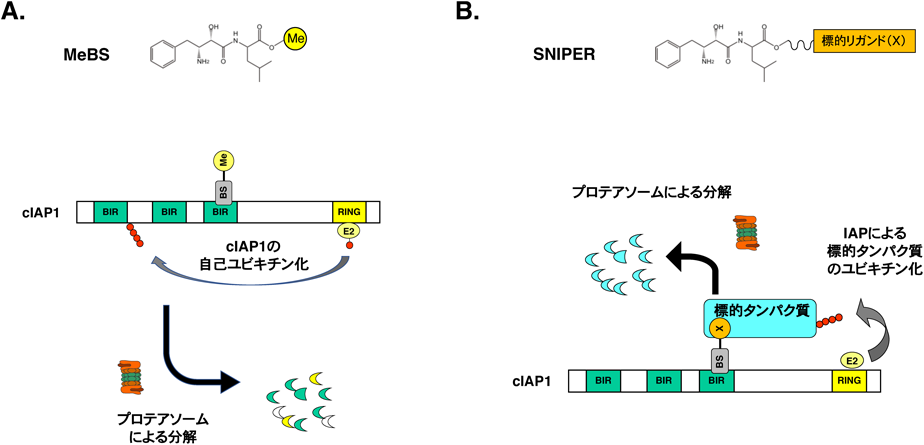
我々の研究室では,IAPファミリータンパク質による細胞死制御機構を研究していたが,その過程でメチルベスタチン(methyl-bestatin:MeBS)がIAPファミリータンパク質のcIAP1を特異的に減少させることを見いだした80).cIAP1はE2ユビキチン結合酵素と結合するRINGドメインを持つタンパク質であり,E3としての機能を持っている.詳しい解析の結果,MeBSはcIAP1のBIR3ドメインと相互作用し,cIAP1のRING依存的な自己ユビキチン化を活性化してプロテアソームによる分解を誘導することを明らかにした.またMeBSの構造活性相関解析から,MeBSのメチルエステルを比較的大きな置換基に換えてもこの活性は保持されるが,ベスタチン骨格を修飾するとcIAP1との相互作用が失われ,cIAP1分解誘導活性もなくなることがわかった.したがってMeBSはベスタチン骨格を介してBIR3ドメインと相互作用していると推測された(図3A).またメチル基はcIAP1から離れていると考えられたことから,MeBSのメチル基を標的タンパク質に結合するリガンドと置換した化合物によって,標的タンパク質をcIAP1によってユビキチン化し,プロテアソームで分解できるのではないかと考えた(図3B).
2)第一世代SNIPER化合物の開発
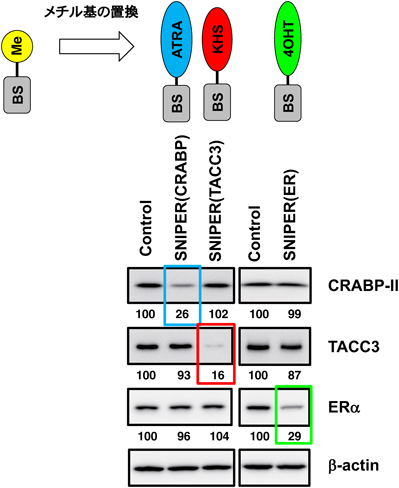
このアイデアを基に,ベスタチンをcIAP1リガンドとして利用した各種SNIPER化合物を産官学の合成化学者とともに共同で開発した.最初に開発したSNIPER(CRABP)はATRA(all-trans retinoic acid)を標的リガンドとして導入した化合物で,ATRA結合タンパク質であるCRABP2(cellular retinoic acid binding protein 2)を減少させる活性を示した54, 81–83).またタモキシフェン(4-hydroxytamoxifen)を標的リガンドとしてエストロゲン受容体(ERα)を分解するSNIPER(ER)を84, 85),さらにKHS-108をリガンドとしてTACC3(transforming acidic coiled-coil)タンパク質を分解するSNIPER(TACC3)を開発した(図4)86).これらの結果から,標的リガンドを取り替えることによって目的とするタンパク質を分解するSNIPER化合物を合理的に開発することができるというコンセプトが実証され,さらにさまざまな標的タンパク質を分解する第一世代SNIPER化合物を開発した87–90).第一世代のSNIPERは標的タンパク質の分解に10 µMまたはそれ以上の濃度を必要とするものが多い.
3)第二世代SNIPERの開発
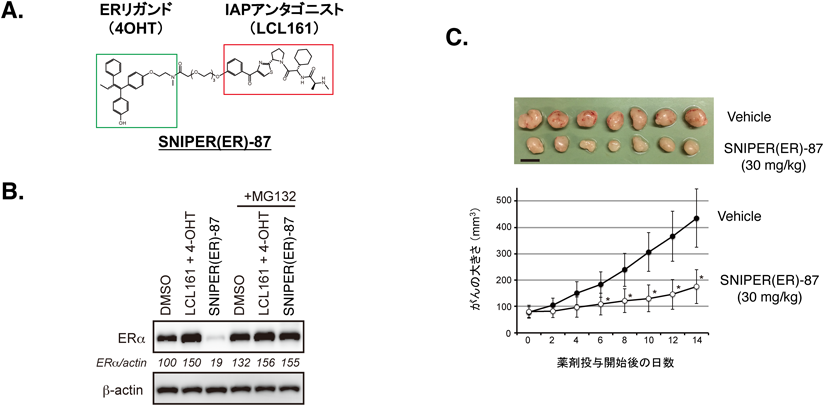
上述したようにIAPファミリータンパク質はがん治療の標的分子として以前から注目されており,多数のIAPアンタゴニストがすでに開発されている.これらのIAPアンタゴニストは,cIAP1だけではなくXIAPなどのBIRドメインにも高い親和性で結合することが報告されている.そこで,これらのIAPアンタゴニストを導入し,より低濃度で標的タンパク質の分解活性を示すSNIPERの開発を試みた.その結果開発されたSNIPER(ER)-87, 105などの化合物は,培養細胞では数十nM程度の濃度でエストロゲン受容体の分解誘導活性を示した(図5A, B).またヒト乳がん細胞を移植したヌードマウスにこれらのSNIPER(ER)を投与すると,がん細胞内のエストロゲン受容体を減少させ,がんの増殖を抑制した(図5C)57, 58).分解誘導メカニズムを詳細に解析した結果,これらのSNIPER(ER)はXIAPを優先的にエストロゲン受容体にリクルートして“エストロゲン受容体-SNIPER-XIAP”の三者複合体を形成することにより,XIAP依存的にエストロゲン受容体をユビキチン化して分解誘導することが明らかになった.また標的リガンドを取り替えることによってBCR-ABL, BRD4, PDE4などの標的タンパク質を数nM~数十nMで分解誘導する各種SNIPER化合物を開発することができた57, 91, 92).これらの結果から,適切な標的リガンドとIAPリガンドを組み合わせることによって,培養細胞では数十nMオーダーという低濃度で標的タンパク質を分解し,in vivoでも分解活性を示すSNIPER化合物を開発するプラットフォーム技術がひとまず完成した.
標的タンパク質の分解を作用機序とするPROTAC/SNIPERなどの化合物は,標的タンパク質の機能を阻害する阻害剤とは異なる薬理学的特徴を示す.化合物のカテゴリーによって多少の違いはあるが,ここでは創薬への応用が進むPROTAC/SNIPERの特徴について主に述べる.
1)触媒的作用
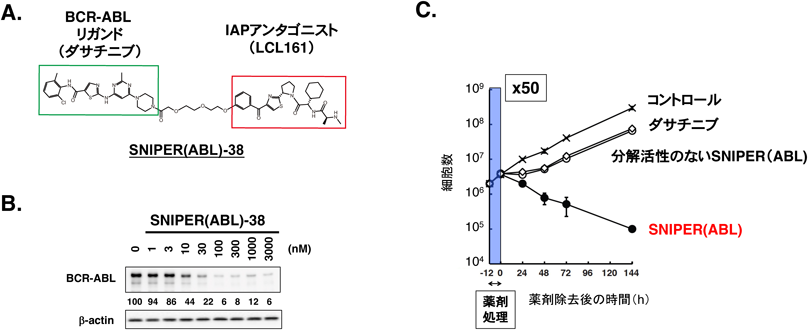
PROTAC/SNIPERは細胞内で三者複合体(E3/PROTAC/標的タンパク質)を形成し,標的タンパク質のユビキチン化と分解を引き起こす.最初の標的タンパク質が分解されると,次の標的タンパク質と複合体を形成することができるため,触媒的に次々と標的タンパク質を分解する62).そのため薬剤濃度が低くても標的タンパク質を分解すると考えられており,これまでに開発されたPROTAC/SNIPERではDC50値(標的タンパク質の量を半減させる濃度)がpMオーダーという非常に低濃度で分解活性を示すものも報告されている93).また単回投与で時間が経過した後も細胞内に残った少量のPROTAC/SNIPERが分解活性を示すため,長時間にわたって標的タンパク質の機能を抑制できる.実際BCR-ABLを分解するSNIPER(ABL)(図6A, B)を慢性骨髄性白血病細胞に12時間処理すると,薬剤を除去した後も白血病細胞の増殖を強く阻害しほとんどの細胞がアポトーシスを起こして死滅したが,キナーゼ阻害剤ダサチニブで同様な実験を行うと白血病細胞は薬剤除去後速やかに再増殖した(図6C)94).このようなPROTAC/SNIPERの特徴は薬理効果の持続性という点では有利な性質であるが,標的タンパク質が分解された結果有害事象が起こった場合にはこれを制御しにくいことが懸念されるため,臨床応用においては十分な注意が必要である.
2)標的タンパク質のすべての機能の抑制
タンパク質は一般に多機能であり,一つのタンパク質がさまざまなタンパク質との相互作用により多様な機能を示すことが多い.このような多機能タンパク質の機能をすべて阻害することは小分子阻害剤では実質的に不可能であるが,標的タンパク質を分解するPROTAC/SNIPERでは可能である.一つのタンパク質が多様な機能で疾患に関与する場合,PROTAC/SNIPERなどのタンパク質分解医薬品を開発することは合理的な戦略の一つと考えられる.
3)標的タンパク質に対する高い特異性
PROTAC/SNIPERが標的タンパク質を分解するためには,三者複合体を形成することが必要であるが,三者複合体を形成すれば必ず分解されるわけではない95).標的タンパク質を分解するためにはユビキチン化が必要であり,そのためにはE3からユビキチンを受け取るリシン残基が標的タンパク質の適切な位置に存在することが必要である.したがって標的リガンドが結合するタンパク質が複数あったとしてもその中で分解されるのは一部の種類に限られ,標的に対する特異性の高いPROTAC/SNIPERを開発することが可能になると考えられている.これまでに開発されたPROTAC/SNIPERの中には,高深度プロテオーム解析で8000以上のタンパク質を精密に比較解析した結果,一つの標的タンパク質だけが分解された例もある93).
4)PROTACとSNIPERの違い
PROTAC/SNIPERでよく利用されるE3と標的タンパク質との複合体を模式的に示した(図7).標的タンパク質はこれらの複合体を介してユビキチン化を受けプロテアソームで分解される.しかし標的タンパク質にリクルートするE3を変えると,標的タンパク質の分解活性が大きく変わることもよくある96, 97).したがって優れた分解活性を持つPROTAC/SNIPERの開発には,標的タンパク質とE3の最適な組み合わせを見いだすことが重要である.
SNIPERはPROTACと異なり標的タンパク質だけでなくE3として働くcIAP1も分解する.そのためE3の減少がSNIPERによる標的タンパク質の分解活性の低下につながる可能性が指摘されることもあるが,細胞内にはXIAPなどSNIPERでは分解されにくいIAPも存在するため標的タンパク質の分解に大きな問題はない.むしろ前述したようにIAPは多くのがん細胞で過剰発現が認められ治療抵抗性に関与することから,XIAPを利用して標的タンパク質を分解しさらにcIAP1を分解するSNIPERは抗がん剤としての利点があると考えられる13, 14).
5)抗体などの創薬モダリティーとの比較
表1にPROTAC/SNIPERと従来の小分子医薬品,抗体などのバイオ医薬品,今後臨床開発が進むと思われる創薬モダリティーとして核酸医薬品,遺伝子治療製品を比較してその特徴をまとめた.PROTAC/SNIPERには,小分子阻害剤や抗体にはない触媒的な作用があり,持続性に優れた経口薬への展開が可能98, 99)という特徴がある.また標的タンパク質は細胞内タンパク質全般であり,細胞外および細胞膜タンパク質を標的とする抗体とは相補的な関係にある.新しい創薬モダリティーとして核酸医薬,遺伝子治療の実用化も進みつつあるが,それぞれに長所短所がある.あらためていうまでもないが,標的タンパク質およびモダリティーの特性などを考慮して,創薬のストラテジーを考えることが重要であろう.
表1 創薬モダリティーの比較 | 小分子阻害剤 | 抗体 | PROTAC/SNIPER | 核酸医薬 | 遺伝子治療 |
|---|
| 分子量 | <500 | 160,000 | 600~1200 | 5000~10,000 | >3,000,000 |
| 標的タンパク質存在部位(具体例) | 細胞内外(酵素,受容体) | 細胞外,細胞膜(液性因子,受容体) | 細胞内(酵素,受容体,転写因子,スプライシング因子等) | 細胞内外(全タンパク質) | 細胞内外(全タンパク質) |
| 持続性 | △ | ○ | ○ | ◎ | ◎ |
| 触媒的作用 | × | × | ○ | ○ | × |
| 組織へのデリバリー | ○ | ○ | ○ | △ | ○ |
| 経口薬 | ○ | × | ○ | × | × |
PROTAC/SNIPERを利用した基礎研究への応用も進みつつある.ユビキチン修飾はプロテアソームによる分解のシグナルとなるだけでなく,細胞膜受容体の内在化と分解,ミトファジー,細胞内シグナル伝達,DNA修復など細胞のさまざまな機能制御に関与することが知られている.ユビキチン修飾によってこれらの多様な機能が制御される仕組みの解明は重要な研究課題の一つであるが,有力な仮説としてユビキチンコード仮説が提唱されている100).これは,ユビキチン修飾は一様ではなくポリユビキチン鎖の長さやK48, K63, K11などのユビキチン鎖の連結様式,さらにはアセチル化やリン酸化などの修飾が組み合わされた複雑なコードをなしており101, 102),そのコードを創る分子,読み解く分子,消去する分子などによってさまざまな細胞機能が制御されるという仮説である.
PROTACの一種MZ1によって標的タンパク質BRD4に形成されるユビキチンコードに関しては,CRL2VHLによるK48ユビキチン鎖に加えて,TRIP12によるK29分岐型ユビキチン鎖が形成され,標的タンパク質の分解が効率よく進むことが明らかになった103).またSNIPERによってミトコンドリアのhexokinase-1あるいはTOMM20タンパク質をユビキチン化するとミトファジーが誘導され104),ミトコンドリア膜上に形成されるユビキチン鎖が単独でもミトファジーを誘導するシグナルとなっていることが示された.このようにPROTAC/SNIPERなどの化合物はユビキチンの機能やユビキチンコードを解析するツールとしての利用も見込まれる.
引用文献References
1) Chabner, B.A. & Roberts, T.G. Jr. (2005) Timeline: Chemotherapy and the war on cancer. Nat. Rev. Cancer, 5, 65–72.
2) Shawver, L.K., Slamon, D., & Ullrich, A. (2002) Smart drugs: Tyrosine kinase inhibitors in cancer therapy. Cancer Cell, 1, 117–123.
3) Noble, M.E., Endicott, J.A., & Johnson, L.N. (2004) Protein kinase inhibitors: Insights into drug design from structure. Science, 303, 1800–1805.
4) Druker, B.J., Talpaz, M., Resta, D.J., Peng, B., Buchdunger, E., Ford, J.M., Lydon, N.B., Kantarjian, H., Capdeville, R., Ohno-Jones, S., et al. (2001) Efficacy and safety of a specific inhibitor of the BCR-ABL tyrosine kinase in chronic myeloid leukemia. N. Engl. J. Med., 344, 1031–1037.
5) Druker, B.J. & Lydon, N.B. (2000) Lessons learned from the development of an abl tyrosine kinase inhibitor for chronic myelogenous leukemia. J. Clin. Invest., 105, 3–7.
6) Carroll, M., Ohno-Jones, S., Tamura, S., Buchdunger, E., Zimmermann, J., Lydon, N.B., Gilliland, D.G., & Druker, B.J. (1997) CGP 57148, a tyrosine kinase inhibitor, inhibits the growth of cells expressing BCR-ABL, TEL-ABL, and TEL-PDGFR fusion proteins. Blood, 90, 4947–4952.
7) Bond, M.J., Chu, L., Nalawansha, D.A., Li, K., & Crews, C.M. (2020) Targeted degradation of oncogenic KRASG12C by VHL-recruiting PROTACs. ACS Cent. Sci., 6, 1367–1375.
8) Buckley, D.L. & Crews, C.M. (2014) Small-molecule control of intracellular protein levels through modulation of the ubiquitin proteasome system. Angew. Chem. Int. Ed. Engl., 53, 2312–2330.
9) Burslem, G.M., Smith, B.E., Lai, A.C., Jaime-Figueroa, S., McQuaid, D.C., Bondeson, D.P., Toure, M., Dong, H., Qian, Y., Wang, J., et al. (2018) The advantages of targeted protein degradation over inhibition: An RTK case study. Cell Chem. Biol., 25, 67–77.e3.
10) Ding, Y., Fei, Y., & Lu, B. (2020) Emerging new concepts of degrader technologies. Trends Pharmacol. Sci., 41, 464–474.
11) Ishikawa, M., Tomoshige, S., Demizu, Y., & Naito, M. (2020) Selective degradation of target proteins by chimeric small-molecular drugs, PROTACs and SNIPERs. Pharmaceuticals (Basel), 13, 74.
12) Maniaci, C., Hughes, S.J., Testa, A., Chen, W., Lamont, D.J., Rocha, S., Alessi, D.R., Romeo, R., & Ciulli, A. (2017) Homo-PROTACs: Bivalent small-molecule dimerizers of the VHL E3 ubiquitin ligase to induce self-degradation. Nat. Commun., 8, 830.
13) Naito, M., Ohoka, N., & Shibata, N. (2019) SNIPERs-Hijacking IAP activity to induce protein degradation. Drug Discov. Today. Technol., 31, 35–42.
14) Naito, M., Ohoka, N., Shibata, N., & Tsukumo, Y. (2019) Targeted protein degradation by chimeric small molecules, PROTACs and SNIPERs. Front Chem., 7, 849.
15) Posternak, G., Tang, X., Maisonneuve, P., Jin, T., Lavoie, H., Daou, S., Orlicky, S., Goullet de Rugy, T., Caldwell, L., Chan, K., et al. (2020) Functional characterization of a PROTAC directed against BRAF mutant V600E. Nat. Chem. Biol., 16, 1170–1178.
16) Raina, K., Lu, J., Qian, Y., Altieri, M., Gordon, D., Rossi, A.M., Wang, J., Chen, X., Dong, H., Siu, K., et al. (2016) PROTAC-induced BET protein degradation as a therapy for castration-resistant prostate cancer. Proc. Natl. Acad. Sci. USA, 113, 7124–7129.
17) Zeng, M., Xiong, Y., Safaee, N., Nowak, R.P., Donovan, K.A., Yuan, C.J., Nabet, B., Gero, T.W., Feru, F., Li, L., et al. (2020) Exploring targeted degradation strategy for oncogenic KRASG12C. Cell Chem. Biol., 27, 19–31.e6.
18) Tanaka, K. (2009) The proteasome: Overview of structure and functions. Proc. Jpn. Acad., Ser. B, Phys. Biol. Sci., 85, 12–36.
19) Ravid, T. & Hochstrasser, M. (2008) Diversity of degradation signals in the ubiquitin-proteasome system. Nat. Rev. Mol. Cell Biol., 9, 679–690.
20) Ciechanover, A. (1994) The ubiquitin-proteasome proteolytic pathway. Cell, 79, 13–21.
21) Ciechanover, A. (1998) The ubiquitin-proteasome pathway: On protein death and cell life. EMBO J., 17, 7151–7160.
22) Yim, W.W. & Mizushima, N. (2020) Lysosome biology in autophagy. Cell Discov., 6, 6.
23) Bonam, S.R., Wang, F., & Muller, S. (2019) Lysosomes as a therapeutic target. Nat. Rev. Drug Discov., 18, 923–948.
24) Nishimura, K., Fukagawa, T., Takisawa, H., Kakimoto, T., & Kanemaki, M. (2009) An auxin-based degron system for the rapid depletion of proteins in nonplant cells. Nat. Methods, 6, 917–922.
25) Natsume, T., Kiyomitsu, T., Saga, Y., & Kanemaki, M.T. (2016) Rapid protein depletion in human cells by auxin-inducible degron tagging with short homology donors. Cell Rep., 15, 210–218.
26) Nabet, B., Roberts, J.M., Buckley, D.L., Paulk, J., Dastjerdi, S., Yang, A., Leggett, A.L., Erb, M.A., Lawlor, M.A., Souza, A., et al. (2018) The dTAG system for immediate and target-specific protein degradation. Nat. Chem. Biol., 14, 431–441.
27) Kepinski, S. & Leyser, O. (2005) The Arabidopsis F-box protein TIR1 is an auxin receptor. Nature, 435, 446–451.
28) Dharmasiri, N., Dharmasiri, S., & Estelle, M. (2005) The F-box protein TIR1 is an auxin receptor. Nature, 435, 441–445.
29) Ito, T., Ando, H., Suzuki, T., Ogura, T., Hotta, K., Imamura, Y., Yamaguchi, Y., & Handa, H. (2010) Identification of a primary target of thalidomide teratogenicity. Science, 327, 1345–1350.
30) Lu, G., Middleton, R.E., Sun, H., Naniong, M., Ott, C.J., Mitsiades, C.S., Wong, K.K., Bradner, J.E., & Kaelin, W.G. Jr. (2014) The myeloma drug lenalidomide promotes the cereblon-dependent destruction of Ikaros proteins. Science, 343, 305–309.
31) Kronke, J., Fink, E.C., Hollenbach, P.W., MacBeth, K.J., Hurst, S.N., Udeshi, N.D., Chamberlain, P.P., Mani, D.R., Man, H.W., Gandhi, A.K., et al. (2015) Lenalidomide induces ubiquitination and degradation of CK1α in del(5q) MDS. Nature, 523, 183–188.
32) Kronke, J., Udeshi, N.D., Narla, A., Grauman, P., Hurst, S.N., McConkey, M., Svinkina, T., Heckl, D., Comer, E., Li, X., et al. (2014) Lenalidomide causes selective degradation of IKZF1 and IKZF3 in multiple myeloma cells. Science, 343, 301–305.
33) Sievers, Q.L., Petzold, G., Bunker, R.D., Renneville, A., Slabicki, M., Liddicoat, B.J., Abdulrahman, W., Mikkelsen, T., Ebert, B.L., & Thoma, N.H. (2018). Science, 362, eaat0572.
34) Matyskiela, M.E., Lu, G., Ito, T., Pagarigan, B., Lu, C.C., Miller, K., Fang, W., Wang, N.Y., Nguyen, D., Houston, J., et al. (2016) A novel cereblon modulator recruits GSPT1 to the CRL4(CRBN) ubiquitin ligase. Nature, 535, 252–257.
35) Uehara, T., Minoshima, Y., Sagane, K., Sugi, N.H., Mitsuhashi, K.O., Yamamoto, N., Kamiyama, H., Takahashi, K., Kotake, Y., Uesugi, M., et al. (2017) Selective degradation of splicing factor CAPERα by anticancer sulfonamides. Nat. Chem. Biol., 13, 675–680.
36) Han, T., Goralski, M., Gaskill, N., Capota, E., Kim, J., Ting, T.C., Xie, Y., Williams, N.S., & Nijhawan, D. (2017). Science, 356, eaal3755.
37) Slabicki, M., Kozicka, Z., Petzold, G., Li, Y.D., Manojkumar, M., Bunker, R.D., Donovan, K.A., Sievers, Q.L., Koeppel, J., Suchyta, D., et al. (2020) The CDK inhibitor CR8 acts as a molecular glue degrader that depletes cyclin K. Nature, 585, 293–297.
38) Slabicki, M., Yoon, H., Koeppel, J., Nitsch, L., Roy Burman, S.S., Di Genua, C., Donovan, K.A., Sperling, A.S., Hunkeler, M., Tsai, J.M., et al. (2020) Small-molecule-induced polymerization triggers degradation of BCL6. Nature, 588, 164–168.
39) Wittmann, B.M., Sherk, A., & McDonnell, D.P. (2007) Definition of functionally important mechanistic differences among selective estrogen receptor down-regulators. Cancer Res., 67, 9549–9560.
40) DeFriend, D.J., Howell, A., Nicholson, R.I., Anderson, E., Dowsett, M., Mansel, R.E., Blamey, R.W., Bundred, N.J., Robertson, J.F., Saunders, C., et al. (1994) Investigation of a new pure antiestrogen (ICI 182780) in women with primary breast cancer. Cancer Res., 54, 408–414.
41) Shoda, T., Ohoka, N., Tsuji, G., Fujisato, T., Inoue, H., Demizu, Y., Naito, M., & Kurihara, M. (2020). Pharmaceuticals (Basel), 13, ph13030034.
42) Neklesa, T.K., Tae, H.S., Schneekloth, A.R., Stulberg, M.J., Corson, T.W., Sundberg, T.B., Raina, K., Holley, S.A., & Crews, C.M. (2011) Small-molecule hydrophobic tagging-induced degradation of HaloTag fusion proteins. Nat. Chem. Biol., 7, 538–543.
43) Long, M.J., Gollapalli, D.R., & Hedstrom, L. (2012) Inhibitor mediated protein degradation. Chem. Biol., 19, 629–637.
44) Asawa, Y., Nishida, K., Kawai, K., Domae, K., Ban, H.S., Kitazaki, A., Asami, H., Kohno, J.Y., Okada, S., Tokuma, H., et al. (2021) Carborane as an alternative efficient hydrophobic tag for protein degradation. Bioconjug. Chem., 32, 2377–2385.
45) Fulda, S. & Vucic, D. (2012) Targeting IAP proteins for therapeutic intervention in cancer. Nat. Rev. Drug Discov., 11, 109–124.
46) Pedersen, J., LaCasse, E.C., Seidelin, J.B., Coskun, M., & Nielsen, O.H. (2014) Inhibitors of apoptosis (IAPs) regulate intestinal immunity and inflammatory bowel disease (IBD) inflammation. Trends Mol. Med., 20, 652–665.
47) Varfolomeev, E., Blankenship, J.W., Wayson, S.M., Fedorova, A.V., Kayagaki, N., Garg, P., Zobel, K., Dynek, J.N., Elliott, L.O., Wallweber, H.J., et al. (2007) IAP antagonists induce autoubiquitination of c-IAPs, NF-kappaB activation, and TNFalpha-dependent apoptosis. Cell, 131, 669–681.
48) Vince, J.E., Wong, W.W., Khan, N., Feltham, R., Chau, D., Ahmed, A.U., Benetatos, C.A., Chunduru, S.K., Condon, S.M., McKinlay, M., et al. (2007) IAP antagonists target cIAP1 to induce TNFalpha-dependent apoptosis. Cell, 131, 682–693.
49) Tamanini, E., Buck, I.M., Chessari, G., Chiarparin, E., Day, J.E.H., Frederickson, M., Griffiths-Jones, C.M., Hearn, K., Heightman, T.D., Iqbal, A., et al. (2017) Discovery of a potent nonpeptidomimetic, small-molecule antagonist of cellular inhibitor of apoptosis protein 1 (cIAP1) and X-linked inhibitor of apoptosis protein (XIAP). J. Med. Chem., 60, 4611–4625.
50) Petersen, S.L., Wang, L., Yalcin-Chin, A., Li, L., Peyton, M., Minna, J., Harran, P., & Wang, X. (2007) Autocrine TNFalpha signaling renders human cancer cells susceptible to Smac-mimetic-induced apoptosis. Cancer Cell, 12, 445–456.
51) Li, L., Thomas, R.M., Suzuki, H., De Brabander, J.K., Wang, X., & Harran, P.G. (2004) A small molecule Smac mimic potentiates TRAIL- and TNFalpha-mediated cell death. Science, 305, 1471–1474.
52) Flygare, J.A., Beresini, M., Budha, N., Chan, H., Chan, I.T., Cheeti, S., Cohen, F., Deshayes, K., Doerner, K., Eckhardt, S.G., et al. (2012) Discovery of a potent small-molecule antagonist of inhibitor of apoptosis (IAP) proteins and clinical candidate for the treatment of cancer (GDC-0152). J. Med. Chem., 55, 4101–4113.
53) Sakamoto, K.M., Kim, K.B., Kumagai, A., Mercurio, F., Crews, C.M., & Deshaies, R.J. (2001) Protacs: Chimeric molecules that target proteins to the Skp1-Cullin-F box complex for ubiquitination and degradation. Proc. Natl. Acad. Sci. USA, 98, 8554–8559.
54) Itoh, Y., Ishikawa, M., Naito, M., & Hashimoto, Y. (2010) Protein knockdown using methyl bestatin-ligand hybrid molecules: Design and synthesis of inducers of ubiquitination-mediated degradation of cellular retinoic acid-binding proteins. J. Am. Chem. Soc., 132, 5820–5826.
55) Schneekloth, A.R., Pucheault, M., Tae, H.S., & Crews, C.M. (2008) Targeted intracellular protein degradation induced by a small molecule: En route to chemical proteomics. Bioorg. Med. Chem. Lett., 18, 5904–5908.
56) Zengerle, M., Chan, K.H., & Ciulli, A. (2015) Selective small molecule induced degradation of the BET bromodomain protein BRD4. ACS Chem. Biol., 10, 1770–1777.
57) Ohoka, N., Okuhira, K., Ito, M., Nagai, K., Shibata, N., Hattori, T., Ujikawa, O., Shimokawa, K., Sano, O., Koyama, R., et al. (2017) In Vivo knockdown of pathogenic proteins via Specific and Nongenetic Inhibitor of Apoptosis Protein (IAP)-dependent Protein Erasers (SNIPERs). J. Biol. Chem., 292, 4556–4570.
58) Ohoka, N., Morita, Y., Nagai, K., Shimokawa, K., Ujikawa, O., Fujimori, I., Ito, M., Hayase, Y., Okuhira, K., Shibata, N., et al. (2018) Derivatization of inhibitor of apoptosis protein (IAP) ligands yields improved inducers of estrogen receptor α degradation. J. Biol. Chem., 293, 6776–6790.
59) Zorba, A., Nguyen, C., Xu, Y., Starr, J., Borzilleri, K., Smith, J., Zhu, H., Farley, K.A., Ding, W., Schiemer, J., et al. (2018) Delineating the role of cooperativity in the design of potent PROTACs for BTK. Proc. Natl. Acad. Sci. USA, 115, E7285–E7292.
60) Winter, G.E., Buckley, D.L., Paulk, J., Roberts, J.M., Souza, A., Dhe-Paganon, S., & Bradner, J.E. (2015) DRUG DEVELOPMENT. Phthalimide conjugation as a strategy for in vivo target protein degradation. Science, 348, 1376–1381.
61) Burslem, G.M., Song, J., Chen, X., Hines, J., & Crews, C.M. (2018) Enhancing antiproliferative activity and selectivity of a FLT-3 inhibitor by proteolysis targeting chimera conversion. J. Am. Chem. Soc., 140, 16428–16432.
62) Bondeson, D.P., Mares, A., Smith, I.E., Ko, E., Campos, S., Miah, A.H., Mulholland, K.E., Routly, N., Buckley, D.L., Gustafson, J.L., et al. (2015) Catalytic in vivo protein knockdown by small-molecule PROTACs. Nat. Chem. Biol., 11, 611–617.
63) Deshaies, R.J. (2015) Protein degradation: Prime time for PROTACs. Nat. Chem. Biol., 11, 634–635.
64) Zhang, X., Crowley, V.M., Wucherpfennig, T.G., Dix, M.M., & Cravatt, B.F. (2019) Electrophilic PROTACs that degrade nuclear proteins by engaging DCAF16. Nat. Chem. Biol., 15, 737–746.
65) Ward, C.C., Kleinman, J.I., Brittain, S.M., Lee, P.S., Chung, C.Y.S., Kim, K., Petri, Y., Thomas, J.R., Tallarico, J.A., McKenna, J.M., et al. (2019) Covalent ligand screening uncovers a RNF4 E3 ligase recruiter for targeted protein degradation applications. ACS Chem. Biol., 14, 2430–2440.
66) Spradlin, J.N., Hu, X., Ward, C.C., Brittain, S.M., Jones, M.D., Ou, L., To, M., Proudfoot, A., Ornelas, E., Woldegiorgis, M., et al. (2019) Harnessing the anti-cancer natural product nimbolide for targeted protein degradation. Nat. Chem. Biol., 15, 747–755.
67) Ohoka, N., Tsuji, G., Shoda, T., Fujisato, T., Kurihara, M., Demizu, Y., & Naito, M. (2019) Development of small molecule chimeras that recruit AhR E3 ligase to target proteins. ACS Chem. Biol., 14, 2822–2832.
68) Lu, M., Liu, T., Jiao, Q., Ji, J., Tao, M., Liu, Y., You, Q., & Jiang, Z. (2018) Discovery of a Keap1-dependent peptide PROTAC to knockdown Tau by ubiquitination-proteasome degradation pathway. Eur. J. Med. Chem., 146, 251–259.
69) Hines, J., Lartigue, S., Dong, H., Qian, Y., & Crews, C.M. (2019) MDM2-recruiting PROTAC offers superior, synergistic antiproliferative activity via simultaneous degradation of BRD4 and stabilization of p53. Cancer Res., 79, 251–262.
70) Harrigan, J.A., Jacq, X., Martin, N.M., & Jackson, S.P. (2018) Deubiquitylating enzymes and drug discovery: Emerging opportunities. Nat. Rev. Drug Discov., 17, 57–78.
71) Pal, A., Young, M.A., & Donato, N.J. (2014) Emerging potential of therapeutic targeting of ubiquitin-specific proteases in the treatment of cancer. Cancer Res., 74, 4955–4966.
72) Shibata, N., Ohoka, N., Tsuji, G., Demizu, Y., Miyawaza, K., Ui-Tei, K., Akiyama, T., & Naito, M. (2020) Deubiquitylase USP25 prevents degradation of BCR-ABL protein and ensures proliferation of Ph-positive leukemia cells. Oncogene, 39, 3867–3878.
73) Gierisch, M.E., Pedot, G., Walser, F., Lopez-Garcia, L.A., Jaaks, P., Niggli, F.K., & Schafer, B.W. (2019) USP19 deubiquitinates EWS-FLI1 to regulate Ewing sarcoma growth. Sci. Rep., 9, 951.
74) Wang, S., Kollipara, R.K., Srivastava, N., Li, R., Ravindranathan, P., Hernandez, E., Freeman, E., Humphries, C.G., Kapur, P., Lotan, Y., et al. (2014) Ablation of the oncogenic transcription factor ERG by deubiquitinase inhibition in prostate cancer. Proc. Natl. Acad. Sci. USA, 111, 4251–4256.
75) Takahashi, D. & Arimoto, H. (2020) Targeting selective autophagy by AUTAC degraders. Autophagy, 16, 765–766.
76) Takahashi, D., Moriyama, J., Nakamura, T., Miki, E., Takahashi, E., Sato, A., Akaike, T., Itto-Nakama, K., & Arimoto, H. (2019) AUTACs: Cargo-specific degraders using selective autophagy. Mol. Cell, 76, 797–810.e10.
77) Banik, S.M., Pedram, K., Wisnovsky, S., Ahn, G., Riley, N.M., & Bertozzi, C.R. (2020) Lysosome-targeting chimaeras for degradation of extracellular proteins. Nature, 584, 291–297.
78) Li, Z., Wang, C., Wang, Z., Zhu, C., Li, J., Sha, T., Ma, L., Gao, C., Yang, Y., Sun, Y., et al. (2019) Allele-selective lowering of mutant HTT protein by HTT-LC3 linker compounds. Nature, 575, 203–209.
79) Li, Z., Zhu, C., Ding, Y., Fei, Y., & Lu, B. (2020) ATTEC: A potential new approach to target proteinopathies. Autophagy, 16, 185–187.
80) Sekine, K., Takubo, K., Kikuchi, R., Nishimoto, M., Kitagawa, M., Abe, F., Nishikawa, K., Tsuruo, T., & Naito, M. (2008) Small molecules destabilize cIAP1 by activating auto-ubiquitylation. J. Biol. Chem., 283, 8961–8968.
81) Okuhira, K., Shoda, T., Omura, R., Ohoka, N., Hattori, T., Shibata, N., Demizu, Y., Sugihara, R., Ichino, A., Kawahara, H., et al. (2017) Targeted degradation of proteins localized in subcellular compartments by hybrid small molecules. Mol. Pharmacol., 91, 159–166.
82) Okuhira, K., Ohoka, N., Sai, K., Nishimaki-Mogami, T., Itoh, Y., Ishikawa, M., Hashimoto, Y., & Naito, M. (2011) Specific degradation of CRABP-II via cIAP1-mediated ubiquitylation induced by hybrid molecules that crosslink cIAP1 and the target protein. FEBS Lett., 585, 1147–1152.
83) Itoh, Y., Ishikawa, M., Kitaguchi, R., Okuhira, K., Naito, M., & Hashimoto, Y. (2012) Double protein knockdown of cIAP1 and CRABP-II using a hybrid molecule consisting of ATRA and IAPs antagonist. Bioorg. Med. Chem. Lett., 22, 4453–4457.
84) Okuhira, K., Demizu, Y., Hattori, T., Ohoka, N., Shibata, N., Nishimaki-Mogami, T., Okuda, H., Kurihara, M., & Naito, M. (2013) Development of hybrid small molecules that induce degradation of estrogen receptor-alpha and necrotic cell death in breast cancer cells. Cancer Sci., 104, 1492–1498.
85) Demizu, Y., Okuhira, K., Motoi, H., Ohno, A., Shoda, T., Fukuhara, K., Okuda, H., Naito, M., & Kurihara, M. (2012) Design and synthesis of estrogen receptor degradation inducer based on a protein knockdown strategy. Bioorg. Med. Chem. Lett., 22, 1793–1796.
86) Ohoka, N., Nagai, K., Hattori, T., Okuhira, K., Shibata, N., Cho, N., & Naito, M. (2014) Cancer cell death induced by novel small molecules degrading the TACC3 protein via the ubiquitin-proteasome pathway. Cell Death Dis., 5, e1513.
87) Tomoshige, S., Naito, M., Hashimoto, Y., & Ishikawa, M. (2015) Degradation of HaloTag-fused nuclear proteins using bestatin-HaloTag ligand hybrid molecules. Org. Biomol. Chem., 13, 9746–9750.
88) Okitsu, K., Hattori, T., Misawa, T., Shoda, T., Kurihara, M., Naito, M., & Demizu, Y. (2018) Development of a small hybrid molecule that mediates degradation of His-tag fused proteins. J. Med. Chem., 61, 576–582.
89) Hattori, T., Okitsu, K., Yamazaki, N., Ohoka, N., Shibata, N., Misawa, T., Kurihara, M., Demizu, Y., & Naito, M. (2017) Simple and efficient knockdown of His-tagged proteins by ternary molecules consisting of a His-tag ligand, a ubiquitin ligase ligand, and a cell-penetrating peptide. Bioorg. Med. Chem. Lett., 27, 4478–4481.
90) Demizu, Y., Shibata, N., Hattori, T., Ohoka, N., Motoi, H., Misawa, T., Shoda, T., Naito, M., & Kurihara, M. (2016) Development of BCR-ABL degradation inducers via the conjugation of an imatinib derivative and a cIAP1 ligand. Bioorg. Med. Chem. Lett., 26, 4865–4869.
91) Shibata, N., Miyamoto, N., Nagai, K., Shimokawa, K., Sameshima, T., Ohoka, N., Hattori, T., Imaeda, Y., Nara, H., Cho, N., et al. (2017) Development of protein degradation inducers of oncogenic BCR-ABL protein by conjugation of ABL kinase inhibitors and IAP ligands. Cancer Sci., 108, 1657–1666.
92) Shibata, N., Nagai, K., Morita, Y., Ujikawa, O., Ohoka, N., Hattori, T., Koyama, R., Sano, O., Imaeda, Y., Nara, H., et al. (2018) Development of protein degradation inducers of androgen receptor by conjugation of androgen receptor ligands and inhibitor of apoptosis protein ligands. J. Med. Chem., 61, 543–575.
93) Yokoo, H., Shibata, N., Endo, A., Ito, T., Yanase, Y., Murakami, Y., Fujii, K., Hamamura, K., Saeki, Y., Naito, M., et al. (2021) Discovery of a highly potent and selective degrader targeting hematopoietic prostaglandin D synthase via in silico design. J. Med. Chem., 64, 15868–15882.
94) Shibata, N., Shimokawa, K., Nagai, K., Ohoka, N., Hattori, T., Miyamoto, N., Ujikawa, O., Sameshima, T., Nara, H., Cho, N., et al. (2018) Pharmacological difference between degrader and inhibitor against oncogenic BCR-ABL kinase. Sci. Rep., 8, 13549.
95) Huang, H.T., Dobrovolsky, D., Paulk, J., Yang, G., Weisberg, E.L., Doctor, Z.M., Buckley, D.L., Cho, J.H., Ko, E., Jang, J., et al. (2018) A chemoproteomic approach to query the degradable kinome using a multi-kinase degrader. Cell Chem. Biol., 25, 88–99.e6.
96) Steinebach, C., Ng, Y.L.D., Sosic, I., Lee, C.S., Chen, S., Lindner, S., Vu, L.P., Bricelj, A., Haschemi, R., Monschke, M., et al. (2020) Systematic exploration of different E3 ubiquitin ligases: An approach towards potent and selective CDK6 degraders. Chem. Sci. (Camb.), 11, 3474–3486.
97) Lai, A.C., Toure, M., Hellerschmied, D., Salami, J., Jaime-Figueroa, S., Ko, E., Hines, J., & Crews, C.M. (2016) Modular PROTAC design for the degradation of oncogenic BCR-ABL. Angew. Chem. Int. Ed. Engl., 55, 807–810.
98) Xiang, W., Zhao, L., Han, X., Qin, C., Miao, B., McEachern, D., Wang, Y., Metwally, H., Kirchhoff, P.D., Wang, L., et al. (2021) Discovery of ARD-2585 as an exceptionally potent and orally active PROTAC degrader of androgen receptor for the treatment of advanced prostate cancer. J. Med. Chem., 64, 13487–13509.
99) Pike, A., Williamson, B., Harlfinger, S., Martin, S., & McGinnity, D.F. (2020) Optimising proteolysis-targeting chimeras (PROTACs) for oral drug delivery: a drug metabolism and pharmacokinetics perspective. Drug Discov. Today, 25, 1793–1800.
100) Komander, D. & Rape, M. (2012) The ubiquitin code. Annu. Rev. Biochem., 81, 203–229.
101) Ohtake, F., Saeki, Y., Ishido, S., Kanno, J., & Tanaka, K. (2016) The K48-K63 branched ubiquitin chain regulates NF-κB signaling. Mol. Cell, 64, 251–266.
102) Ohtake, F., Tsuchiya, H., Saeki, Y., & Tanaka, K. (2018) K63 ubiquitylation triggers proteasomal degradation by seeding branched ubiquitin chains. Proc. Natl. Acad. Sci. USA, 115, E1401–E1408.
103) Kaiho-Soma, A., Akizuki, Y., Igarashi, K., Endo, A., Shoda, T., Kawase, Y., Demizu, Y., Naito, M., Saeki, Y., Tanaka, K., et al. (2021) TRIP12 promotes small-molecule-induced degradation through K29/K48-branched ubiquitin chains. Mol. Cell, 81, 1411–1424.e7.
104) Yamano, K., Kikuchi, R., Kojima, W., Hayashida, R., Koyano, F., Kawawaki, J., Shoda, T., Demizu, Y., Naito, M., Tanaka, K., et al. (2020) Critical role of mitochondrial ubiquitination and the OPTN-ATG9A axis in mitophagy. J. Cell Biol., 219, e201912144.
105) Wang, W., Zhou, Q., Jiang, T., Li, S., Ye, J., Zheng, J., Wang, X., Liu, Y., Deng, M., Ke, D., et al. (2021) A novel small-molecule PROTAC selectively promotes tau clearance to improve cognitive functions in Alzheimer-like models. Theranostics, 11, 5279–5295.
106) Tomoshige, S. & Ishikawa, M. (2021) PROTACs and other chemical protein degradation technologies for the treatment of neurodegenerative disorders. Angew. Chem. Int. Ed. Engl., 60, 3346–3354.
107) Tomoshige, S., Nomura, S., Ohgane, K., Hashimoto, Y., & Ishikawa, M. (2017) Discovery of small molecules that induce the degradation of huntingtin. Angew. Chem. Int. Ed. Engl., 56, 11530–11533.
108) Silva, M.C., Ferguson, F.M., Cai, Q., Donovan, K.A., Nandi, G., Patnaik, D., Zhang, T., Huang, H.T., Lucente, D.E., Dickerson, B.C., et al. (2019). eLife, 8, 45457.
109) Yokoo, H., Ohoka, N., Naito, M., & Demizu, Y. (2020) Design and synthesis of peptide-based chimeric molecules to induce degradation of the estrogen and androgen receptors. Bioorg. Med. Chem., 28, 115595.
110) Ohoka, N., Misawa, T., Kurihara, M., Demizu, Y., & Naito, M. (2017) Development of a peptide-based inducer of protein degradation targeting NOTCH1. Bioorg. Med. Chem. Lett., 27, 4985–4988.
111) Demizu, Y., Ohoka, N., Nagakubo, T., Yamashita, H., Misawa, T., Okuhira, K., Naito, M., & Kurihara, M. (2016) Development of a peptide-based inducer of nuclear receptors degradation. Bioorg. Med. Chem. Lett., 26, 2655–2658.
112) Shao, J., Yan, Y., Ding, D., Wang, D., He, Y., Pan, Y., Yan, W., Kharbanda, A., Li, H.Y., & Huang, H. (2021) Destruction of DNA-binding proteins by programmable oligonucleotide PROTAC (O’PROTAC): Effective targeting of LEF1 and ERG. Adv. Sci. (Weinh.), 8, e2102555.
113) Samarasinghe, K.T.G., Jaime-Figueroa, S., Burgess, M., Nalawansha, D.A., Dai, K., Hu, Z., Bebenek, A., Holley, S.A., & Crews, C.M. (2021) Targeted degradation of transcription factors by TRAFTACs: TRAnscription Factor TArgeting Chimeras. Cell Chem. Biol., 28, 648–661.
114) Liu, J., Chen, H., Kaniskan, H.U., Xie, L., Chen, X., Jin, J., & Wei, W. (2021) TF-PROTACs enable targeted degradation of transcription factors. J. Am. Chem. Soc., 143, 8902–8910.
115) Smith, B.E., Wang, S.L., Jaime-Figueroa, S., Harbin, A., Wang, J., Hamman, B.D., & Crews, C.M. (2019) Differential PROTAC substrate specificity dictated by orientation of recruited E3 ligase. Nat. Commun., 10, 131.