ミトコンドリアは細胞内のエネルギー産生を担う細胞小器官(オルガネラ)であり,脂質二重膜でできた外膜と内膜を持ち,ミトコンドリア内には独自のミトコンドリアDNAを保持している.ミトコンドリア内膜に局在するミトコンドリア呼吸鎖複合体がプロトン濃度勾配による膜電位差を作り出し,その膜電位差を利用してATPが産生される.ミトコンドリアは,ATP産生だけでなく,熱産生,脂質合成,カルシウム濃度調節,細胞死の制御などさまざまな機能を担う.ミトコンドリアの機能障害はがん,神経変性疾患,心疾患,など多くの疾患との関連が知られている.
ミトコンドリアは分裂や融合を頻繁に行う動的なオルガネラであり,細胞の状態によりその形態を変化させる.ミトコンドリアダイナミクスは,ミトコンドリアの品質管理や細胞死など細胞内イベントにも関与しており,ミトコンドリアダイナミクスの破綻はミトコンドリアの機能障害や細胞の脆弱性の亢進などを引き起こす.細胞に過度なストレスがかかると,ミトコンドリアは分裂し,ミトコンドリア内に局在するチトクロムcが細胞質に放出され,細胞死を誘導するアポトソームが活性化され細胞死が誘導される.また,機能不全に陥った不良ミトコンドリアは活性酸素を過剰に産生し,細胞に障害を与える.この不良ミトコンドリアの分解にもミトコンドリアの分裂が関与する.このようにミトコンドリアの分裂は細胞死や不良ミトコンドリアの除去に関与し,ミトコンドリアの機能維持,細胞内の恒常性を保つために重要な働きを担っている.ミトコンドリア分裂はダイナミン様GTPase Drp1がミトコンドリア外膜上に集積し,ミトコンドリア膜を収縮させることにより引き起こされる.Drp1受容体はミトコンドリア外膜に存在しており,ミトコンドリア外膜においてミトコンドリア分裂は制御されている.また,ミトコンドリアは,細胞周期や飢餓刺激によってミトコンドリアどうしの融合が認められる.飢餓刺激によって誘導されるオートファジーは細胞質成分やオルガネラを分解し,アミノ酸などの細胞生存に必要な物質を供給する.飢餓時においてもミトコンドリアによるATP産生が必要であることから,ミトコンドリアは伸長し,大きい形態をとることにより,オートファジーによる分解を回避する1, 2).ミトコンドリア外膜の融合はミトコンドリア外膜貫通型のタンパク質であるMitofusin 1(Mfn1),Mitofusin 2(Mfn2)が担う.二つのミトコンドリアのMfn1/Mfn2が結合し,複合体を形成することにより,二つのミトコンドリア外膜が接着し,距離を縮め,Mfn1/Mfn2のGTPase活性依存的に外膜の融合が起こる3).ミトコンドリアの分裂・融合はミトコンドリア分裂因子・融合因子によって制御されており,ミトコンドリア外膜上における分裂因子・融合因子の制御系が重要である.
ミトコンドリアは小胞体,リソソーム,脂肪滴,メラノソームなどの他のオルガネラと機能的なコンタクトを形成することが次々と報告されている.ミトコンドリアと小胞体の接着(mitochondria-ER contact sites:MERCs)はミトコンドリアの分裂やミトコンドリアと小胞体間での脂質やカルシウムなどの輸送を担い,ミトコンドリア膜構造を構成する脂質の供給,ATP産生や細胞死を制御する4–6).MERCsを介した小胞体からミトコンドリアへの過剰なカルシウムの流入は,ミトコンドリアを介した細胞死の制御と密接な関連がある.ミトコンドリア内のカルシウム濃度の過剰な増加は,voltage-dependent anion-selective channel 1(VDAC1),adenine nucleotide translocase(ANT),F1–Fo ATP合成酵素,cyclophilin-D(CypD)により構成されるミトコンドリア膜透過性遷移孔(mitochondrial permeability transition pore:mPTP)を開き,細胞死を引き起こす7, 8).mPTP開口によりミトコンドリアの膜電位の消失やミトコンドリア膨張が引き起こされた結果,チトクロムcの放出につながると考えられている9).実際に,MERCsのテザリング因子として知られているPDZD8, Mfn2, Fis1-Bap31の発現を抑制すると,アポトーシス誘導刺激時にミトコンドリア内へのカルシウム流入が減弱し,アポトーシスが抑制される5, 10, 11).細胞の異常事態の際に速やかにミトコンドリア内にカルシウムを流入させ,細胞死を誘導させるために,小胞体からミトコンドリアに直接カルシウムを輸送させるMERCsは非常に合理的な機構である.ミトコンドリアからチトクロムcが細胞質に放出されると,チトクロムcはApaf-1とアポトソームと呼ばれる複合体を形成し,Caspaseファミリーの活性化によるアポトーシスを誘起する.複数のミトコンドリア外膜貫通型タンパク質がMERCsのテザリング因子として同定されており,ミトコンドリア外膜が他のオルガネラとの連携で重要な役割を担うことが明らかである.
ミトコンドリア外膜上では,ミトコンドリア分裂・融合,オルガネラ間コンタクト,マイトファジー,炎症応答などさまざまな細胞内イベントが起きている.ミトコンドリア外膜上におけるそれらの細胞内イベントの制御系の詳細な解明は,ミトコンドリアの正常な機能,細胞死,炎症応答などのシグナル伝達の理解に重要である.本稿では,ミトコンドリア外膜貫通型ユビキチンリガーゼMITOL/MARCHF5がミトコンドリアダイナミクス,MERCsの形成制御,炎症応答の制御を介してミトコンドリアの品質管理,細胞内のシグナル伝達を制御することについて概説する.
2. ミトコンドリア外膜貫通型ユビキチンリガーゼMITOL/MARCHF5
ミトコンドリア外膜貫通型ユビキチンリガーゼであるmitochondrial ubiquitin ligase(MITOL)はMARCHF5, MARCH5, RNF153との別名が存在するが,本稿ではMITOLと記載する.MITOLは2004年にmembrane-associated RING-CH(MARCH)ファミリーのメンバーとして同定され,小胞体に局在する分子として初めて報告された12).2006年に,MITOLはミトコンドリアに局在し,ミトコンドリア外膜貫通型タンパク質であり,ユビキチンリガーゼ活性を有するなど重要な知見が報告され13),ミトコンドリア外膜上でミトコンドリアダイナミクス,変性タンパク質の分解,ミトコンドリアと小胞体のコンタクト,炎症応答などさまざまな細胞内の機構に関与することが報告されている.さらにMITOLはアルツハイマー病などの疾患との関連について報告されており,MITOLがミトコンドリア外膜上においてミトコンドリアの品質管理,機能制御に重要な役割を担うことが示唆されている.
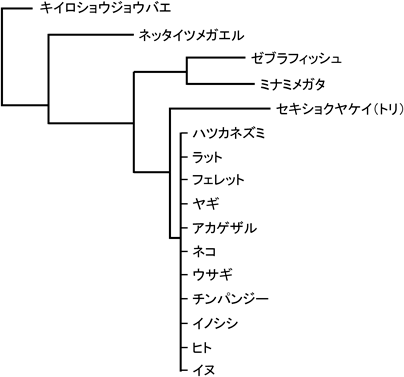
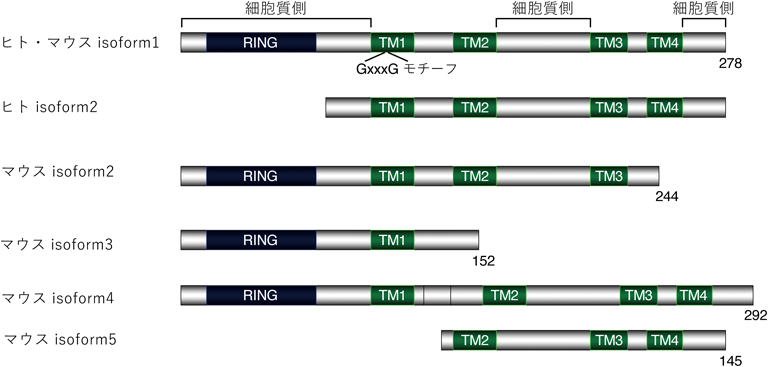
これまでのMITOLに関する研究報告はヒトの培養細胞やマウスを中心に研究されている.NCBIのデータベースによると,MITOLのオルソログはショウジョウバエまで存在する(図1).ショウジョウバエのMITOLとヒトMITOLのタンパク質レベルの相同性は54.9%であるが,ゼブラフィッシュでは97.4%,マウスでは100%の相同性を示し,タンパク質レベルの構造が高度に保存されている(表1).出芽酵母においてMITOLのオルソログは発見されていないが,ゼブラフィッシュから種間で保存されていることから,細胞内の重要なシステム制御系で働くことが予想される.ヒトやマウスのMITOLは278個のアミノ酸からなり,RINGドメインと四つの膜貫通領域(transmembrane domain:TM)を持ち,RINGドメインを含むN末端領域,TM2とTM3に挟まれた領域,C末端領域が細胞質側に露出している(図2).MITOLのRINGドメインに変異を入れた変異体(H43WやC65S/C68S)はユビキチンリガーゼ活性を失うことから13),RINGドメインがMITOLのユビキチンリガーゼ活性の中心を担っている.MITOLはさまざまな基質と結合することが報告されているが,MITOLの基質認識部位としてN末端とC末端の両方が認められている.MITOLは二つの離れたグリシン残基からなるGxxxGモチーフをTM1に持ち,このGxxxGモチーフがMITOLの二量体形成に必要である14).MITOLの二量体形成はMITOL自身の分解に必要であり,MITOLは自身の発現量を調整し,過剰な蓄積による活性化を防いでいる.実際にMITOLの発現プラスミドを細胞に遺伝子導入した際に,MITOLの発現量の劇的な増加は観察されない.一方で,MITOLの発現は細胞種や分化状態によって大きく異なる.脳におけるMITOLの発現量の推移を比較すると,胎生期などの発生期に多いが,成体の脳では少ない15).細胞の状態によってMITOL発現の制御系が異なることから,組織特異的,時期特異的にMITOLの役割が変化すると推察される.
表1 ヒトMITOLとオルソログの配列の比較 | ヒトMITOLとのタンパク質の相同性(%) | ヒトMITOLとのDNAの相同性(%) |
|---|
| チンパンジー | 100 | 100 |
| サル | 100 | 98.8 |
| オオカミ | 99.3 | 95.1 |
| ウシ | 100 | 95.2 |
| マウス | 100 | 92.8 |
| ラット | 100 | 93 |
| セキショクヤケイ(トリ) | 98.2 | 87.2 |
| ネッタイツメガエル | 96.7 | 79.1 |
| ゼブラフィッシュ | 97.4 | 76.8 |
| ショウジョウバエ | 54.9 | 55.6 |
| ガンビエハマダラカ | 49.4 | 53.3 |
| NCBIデータベースに登録されているヒトMITOLと各生物種のMITOLのタンパク質とDNAの相同性の比較. |
MITOLにはスプライシングバリアントが存在するが(図2),isoformの機能的な違いに関する詳細な研究報告はまだされていない.ヒトではRINGドメインを含むN末端領域を欠失するMITOLのヒトisoform2が存在し,このヒトisoform2はユビキチンリガーゼ活性を持たないが,C末端領域の基質認識部位を有することから,基質と結合するがユビキチン化を起こさず,isoform1との拮抗的な役割を担うことが予想される.また,マウスにおいても五つのisoformが存在する.マウスisoform5はヒトisoform2と同様にN末端を欠損している.マウスisoform2とマウスisoform3はRINGドメインを有するが,C末端を欠損しており,C末端領域特異的な基質の認識をしないisoformと考えられる.マウスisoform4はTM1とTM2の間に特異的な配列を有する全長292アミノ酸のisoformであり,isoform1と似た機能を持つことが予想される.
組織や細胞種によってミトコンドリアの形が異なることと同様に,培養細胞やノックアウトマウスを用いた解析においてもMITOLを欠失させた際に基質の蓄積量が細胞種や組織によって異なるという知見が得られている.組織の違いによるMITOL isoformの発現量の違いが,MITOL欠失時の表現型の違いの説明の一つになる可能性があり,MITOLの生理的な役割の解明には,MITOL isoformの詳細な解析やMITOLの組織特異的な役割,MITOLの新たな制御系の解明が必要である.
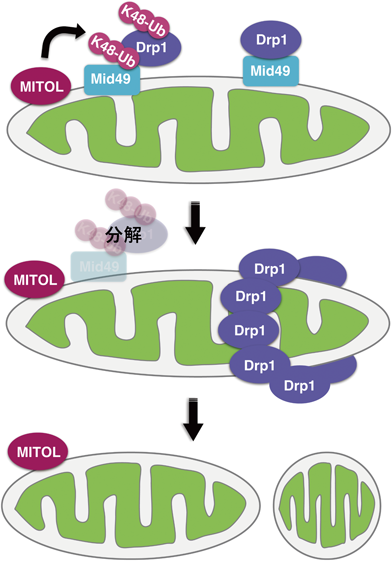
ミトコンドリア分裂は,細胞分裂,細胞の分化,細胞死,マイトファジーなどさまざまな細胞内のイベントに関与する16).ミトコンドリア分裂で中心的な役割を担うのはダイナミン様GTPase Drp1である.Drp1の欠失は,ミトコンドリアの分裂阻害による顕著なミトコンドリアの伸長を示す.Drp1はミトコンドリア外膜に局在するDrp1受容体であるMff, Mid49, Mid51を目印にミトコンドリアにリクルートされる.Drp1は複合体を形成し,ミトコンドリア膜を収縮させ,ミトコンドリア分裂を引き起こす.ミトコンドリア分裂には,小胞体,リソソーム,ゴルジ体由来小胞の関与も報告されている3, 17–19).MITOLはミトコンドリア外膜上でDrp1やMid49のユビキチン化を介してミトコンドリア分裂を制御することが報告されている.
MITOLがミトコンドリアダイナミクスを制御することが初めて報告されたのは2006年である13).MITOLをノックダウンしたHeLa細胞では,分裂状態のミトコンドリアが増加する.MITOLはミトコンドリア分裂因子であるDrp1やhFis1をユビキチン化し,ユビキチン–プロテアソーム依存的な分解を促進する.MITOLの欠失によりDrp1依存的なミトコンドリア分裂が亢進することが示され,MITOLがDrp1依存的なミトコンドリア分裂を抑制することが報告された13).一方,2007年の研究報告では,MITOLのユビキチンリガーゼの変異体であるC65S/C68S, H43Wの過剰発現はDrp1を阻害することによるミトコンドリア分裂阻害を引き起こすことが示され,MITOLがDrp1の活性を正に制御する可能性が示唆された20).相反する機能が報告されたが,現在では,MITOLはDrp1依存的なミトコンドリアの分裂を負に制御することが支持されている.その根拠の一つとして,MITOLがDrp1受容体であるMid49の分解を制御する点である.Drp1は主に細胞質に局在し,ミトコンドリア外膜に局在するMff, Mid49, Mid51といったDrp1受容体によりミトコンドリアにリクルートされ,ミトコンドリア分裂を引き起こす.MITOLはK48型のポリユビキチン鎖をMid49に付加し,分解を促進することによりMid49の発現量を制御する.MITOLを欠損したHCT116細胞では,ミトコンドリアの分裂やMid49の顕著な蓄積が認められている21).過剰なミトコンドリア分裂の誘導はアポトーシス誘導性のストレスに対して脆弱となり,ミトコンドリア分裂の抑制は細胞死を回避する16).MITOL欠損細胞ではMid49が蓄積し,ミトコンドリアの過剰な分裂が引き起こされ,ミトコンドリア呼吸鎖複合体の阻害剤処理などのミトコンドリアに対するストレスに脆弱となる21).ストレス下において,MITOLはミトコンドリアの過剰な分裂を防ぐことで細胞死を調節する役割を担う.興味深いことに,Drp1受容体であるMffとMITOLの結合も認められているが,MITOLはMffの発現量に影響を与えない.MffがMITOLの活性を制御し,MITOLによるMid49の分解を制御することが報告されている22).MITOLを欠損した神経細胞においてもMid49の顕著な蓄積が認められており15),MITOLはMid49/Drp1依存的なミトコンドリア分裂を制御すると考えられる(図3).しかしながら,HeLa細胞やCOS-7細胞に対してsiRNAを用いてMITOLをノックダウンした際には,神経細胞と同様なMid49の顕著な蓄積は認められず,ミトコンドリア分裂におけるMITOLの役割が細胞の状態によって異なる可能性があり,今後新規メカニズムの解明が求められる.
細胞老化の特徴として,細胞の肥大化,テロメアの短縮,活性酸素種の増大,β-ガラクトシダーゼの活性化などが知られている.ミトコンドリアは酸化的リン酸化によるATP合成の際に活性酸素種の発生源となり,細胞老化と密接な関係がある23).ミトコンドリアダイナミクスの破綻はミトコンドリアの品質管理機構に影響を与えることから,細胞老化を促進することが報告されている24).また,鉄キレート剤であるdeferoxamineによって老化を誘導された細胞では,巨大なミトコンドリアが観察されている25).ミトコンドリアダイナミクスを制御するMITOLも老化との関連が示唆されている.MITOLの欠失は老化マーカーの一つであるβ-ガラクトシダーゼ活性の増大を示し,細胞老化を引き起こす26).ミトコンドリア融合活性を持たないMfn1 T109A変異体やDrp1を過剰発現させてミトコンドリアの分裂を促進させると,MITOLの欠失による細胞老化が抑制される.MITOLはMfn1をユビキチン化修飾し,Mfn1の分解を促進し,ミトコンドリアの過剰な融合を阻害する.MITOLの欠失はMfn1による過剰なミトコンドリア融合を引き起こし,細胞老化を亢進させる26).
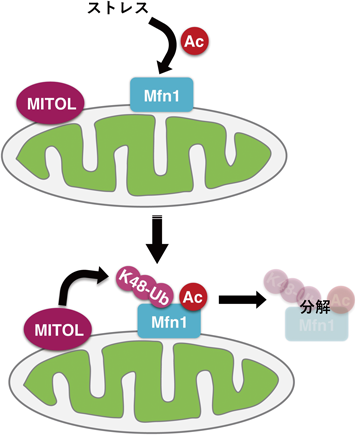
ミトコンドリアは細胞周期によってその形を変化させ,細胞分裂の際に分配されることが知られている.G2/M期において,Drp1はリン酸化され,活性化し,ミトコンドリアを分裂させる27).MITOLはG2/M期においてMfn1を強く認識し,分解を促進することにより,ミトコンドリアの形を制御する28).また,ミトコンドリア呼吸鎖複合体IIIの阻害剤であるantimycin Aで短時間処理すると,Mfn1が蓄積し,ミトコンドリアの伸長が引き起こされる.Mfn1を欠失した細胞にantimycin Aを処理すると細胞死が亢進することから,Mfn1は短時間のミトコンドリアストレスにおいてはミトコンドリアを融合させ,細胞死の抑制に働く.興味深いことに,過剰にMfn1が蓄積した場合においてもミトコンドリアストレスに脆弱となることから,Mfn1の発現量の調節が細胞の生存には重要であり,その役目をMITOLが担っている.antimycin A刺激時にMfn1の491番目のリシンはアセチル化修飾を受ける.MITOLはアセチル化されたMfn1と結合し,ユビキチン化し,分解することにより,Mfn1の発現量を調節し,細胞生存に寄与する29)(図4).このように,細胞分裂時やストレス条件下においてMITOLはMfn1の品質管理を担うことが示されている.
MERCsはカルシウムやリン脂質の効率的な輸送を担っており,さまざまなテザリング因子が局在していることが報告されている.Mfn2はミトコンドリアと小胞体のテザリング因子の一つであり,ミトコンドリアに局在するMfn2と小胞体に局在するMfn2が複合体を形成することにより,MERCsの形成が誘導される5, 30).Mfn2の発現を抑制すると,アポトーシス誘導刺激時に小胞体からミトコンドリアへのカルシウム流入が減弱し,ミトコンドリア内のカルシウム濃度の上昇によるアポトーシスが抑制される.また,Mfn2はリン脂質であるホスファチジルセリンと結合し,MERCsにおけるホスファチジルセリンの輸送を行う31).小胞体で産生されるリン脂質はミトコンドリアの膜構造の形成に必要であり,MERCsを介したリン脂質の輸送はミトコンドリアが正常に機能するために必要である.
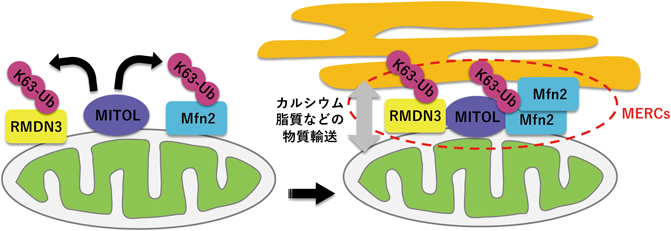
MITOLはミトコンドリア外膜に局在するMfn2と結合する32).MITOLのC末端とMfn2のHR1ドメインがそれぞれの結合領域であり,MITOLがK63型のポリユビキチン化修飾をMfn2に付加する33).K48型のポリユビキチン化修飾とは異なり,K63型のポリユビキチン化修飾されたMfn2はユビキチン-プロテアソームによる分解はされず,Mfn2の複合体形成が誘導される.Mfn2はMfn1と同様にGTPに結合すると,複合体形成が誘導される.MITOLによるMfn2のユビキチン化修飾は,ミトコンドリアと小胞体に局在するMfn2の複合体形成を促進することにより,MERCsの形成を促進する(図5).Mfn2のGTPaseドメイン内にある192番目のリシンがMITOLによるユビキチン修飾部位であり,Mfn2 K192R変異体ではMfn2の複合体形成が阻害される.MITOLを欠失したHeLa細胞では,ミトコンドリアと小胞体の共局在の減少,ミトコンドリアへのカルシウム取り込みの減少などから,MERCsの減少が確認されている33).培養細胞だけでなく,マウスの個体においてもMITOLがMERCsの形成を制御する.連続した電子顕微鏡画像を3次元再構築した解析法により,MITOLを欠損した神経細胞においてMERCsの減少が認められている.MITOLを欠損した脳のミトコンドリアのリン脂質を解析すると,ミトコンドリア内膜の主要なリン脂質であるカルジオリピンの減少が認められた.カルジオリピンは小胞体で産生されるホスファチジン酸からミトコンドリア内で生合成される.ホスファチジン酸はMERCsを介して小胞体からミトコンドリアに運ばれることから,MITOL欠損によるMERCsの減少がミトコンドリア内のカルジオリピンの減少を引き起こしたと推察される.カルジオリピンはミトコンドリア内膜タンパク質の構造維持や活性の調節を担い,ATP産生,酸化ストレス,組織障害と密接な関係がある.実際に,MITOLを欠損した脳では酸化ストレスの増加やグリオーシスの増加が認められ,MITOLを欠損した脳においてカルジオリピンの減少が酸化ストレス増加のトリガーとなったと考えられる.神経細胞においてMITOLはMERCsの形成制御を介してミトコンドリアの品質管理に重要な役割を担うことが示唆された15).
近年,我々は近接タンパク質をビオチン標識するAPEX2を融合させたMITOLを安定的に発現させた細胞を用いて,MITOLの新規結合分子の探索を行った.その結果,これまでに結合が報告されているMfn2, Mfn1, Mffに加えて,新たにRMDN3/PTPIP51をMITOLの基質として同定した.RMDN3はミトコンドリアに局在し,小胞体に局在するVAPBと結合することによりMERCsを形成するテザリング因子である34).RMDN3はホスファチジン酸に結合し,MERCsにおいてホスファチジン酸の輸送に関わることが報告されている35).MITOLはK63型のポリユビキチン鎖をRMDN3に付加することにより,RMDN3のホスファチジン酸結合能を増加させる36).MITOLはMfn2やRMDN3の活性を制御し,MERCsの形成やリン脂質の輸送を制御する(図5).
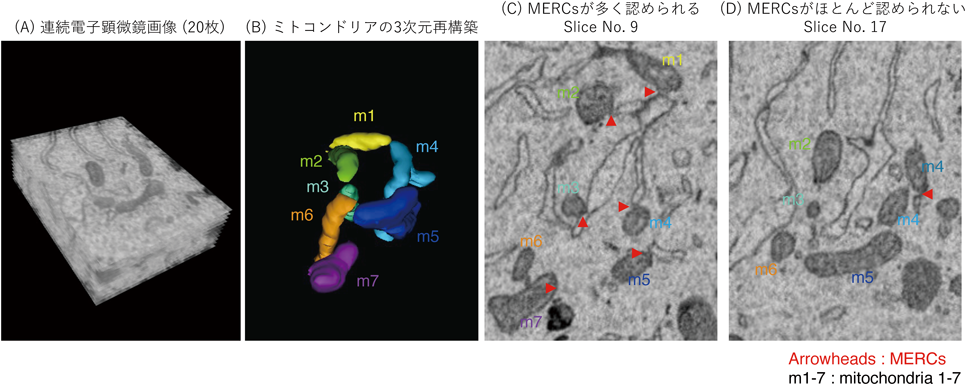
従来のMERCsの解析方法は細胞分画法や2次元的な電子顕微鏡写真を用いた解析が主であり,その解析方法の正確性が不十分であることから,MERCsの実態を正確に捉えることが困難であった.我々は,serial block-face scanning electron microscopy(SBF-SEM)を用いた連続電子顕微鏡画像を3次元再構築した3次元的な解析により,MERCsの面積を正確に定量した(図6)15).この手法は従来の2次元的な解析よりも正確であり,コンタクトの面積を定量できる.図6で示すように同じミトコンドリアでも解析する断面によって小胞体とコンタクトする部位としない部位が存在し,2次元的な解析では断面によりMERCsの数が大きく異なってしまう.実際に同一ミトコンドリアを断面図ごとに観察するとArrowheadsで示したMERCsの数が断面ごとに異なることがわかる(図6).SBF-SEMの電子顕微鏡画像の解像度は13 nm/pixelあり,透過型電子顕微鏡(TEM)画像と比較するとやや劣るが,オルガネラコンタクトの解析には十分である.SBF-SEMを用いて神経細胞におけるミトコンドリアの解析を行うと,同一の細胞であっても細胞内の部位によってミトコンドリアのサイズやMERCsの形成数が異なることが明らかとなった.核に近い細胞体に局在するミトコンドリアは,複数の分岐を持つ複雑な構造を持ち,複数のMERCsが認められるのに対し,神経突起内のミトコンドリアは小さく,MERCsを持たないものが多く認められた.ミトコンドリアの体積とMERCsの数は正の相関が認められることから,小胞体からミトコンドリアへのMERCsを介したリン脂質などの物質輸送がミトコンドリアのサイズを大きくするのに必要であると考えられる.神経突起内では,小さいサイズのミトコンドリアが突起内の限られたスペースを移動するのに有利なためMERCs形成の必要性が低いと推察される.SBF-SEMを用いた3次元的な解析では,各々のMERCsの面積を定量できる大きな利点がある.MERCsの役割は,ミトコンドリア分裂の制御,リン脂質やカルシウムなどの物質輸送がある.ミトコンドリア分裂の際に小胞体がミトコンドリアに巻きつき,ミトコンドリアの分裂を促すことが報告されているが,ミトコンドリアの表面積(周囲長)あたりのMERCsの面積(周囲長)を解析すれば,MERCsあたりのミトコンドリア分裂に関与するMERCsの割合を算出でき,MERCsの形態学的な違いから,MERCsの役割を分類できる可能性があり,3次元的な解析は大きな可能性を秘めている.今後,ミトコンドリアやMERCsのトレースの自動化が進み,広くこの手法が研究に用いられることにより,MERCsだけでなく,ミトコンドリアとペルオキシソーム,ゴルジ体,脂肪滴などさまざまなオルガネラコンタクトの構造的な違いと機能をつなげる研究成果が得られることを期待する.
小胞体は,脂質やステロイドの合成,タンパク質の成熟,カルシウムの貯蔵などを担うオルガネラである.小胞体内にタンパク質が正常に折りたたまれずに蓄積すると小胞体ストレスとなり,小胞体ストレス応答が引き起こされる.小胞体ストレス応答により,タンパク質合成の抑制,シャペロンタンパク質の転写誘導,変性タンパク質の分解除去が誘導される.持続的または重度の小胞体ストレス条件下では,小胞体ストレス応答により細胞死が誘導される37, 38).小胞体ストレス応答を介したアポトーシスは,糖尿病,心不全,神経変性疾患などの病気の原因として知られている39).小胞体はMERCsを介してミトコンドリアの形態制御やカルシウムの流入による細胞死の制御に関与する.ミトコンドリア外膜のタンパク質もMERCsにおいて小胞体ストレス応答などのイベントを制御する可能性が考えられる.
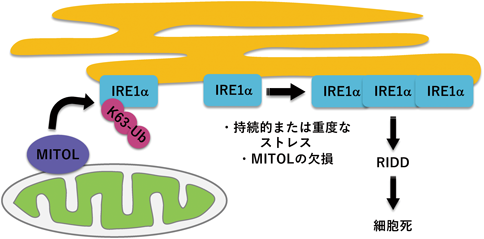
MITOLはミトコンドリア外膜貫通型タンパク質であり,MERCsに多く局在することが示唆されている.MITOL欠損細胞では小胞体の形態変化が認められており33),MITOLが小胞体を制御する可能性が示唆されていた.そこで,小胞体ストレスに対するMITOLの役割の解析を行った.MITOLを欠失した細胞は小胞体の形態変化や小胞体ストレス誘導剤であるthapsigargin, tunicamycin, brefeldin A刺激に対して脆弱であり,MITOLは小胞体ストレス依存的な細胞死に対して細胞保護に働く.小胞体ストレスを感知する3種類のストレスセンサータンパク質(IRE1α, PERK, ATF6)が存在する.MITOLの欠失は,PERK経路とATF6経路に影響を与えないが,IRE1α経路の活性化を介した細胞死の亢進が認められた.実際にIRE1αを抑制したMITOL欠損細胞は,tunicamycin刺激によって誘発される細胞死への脆弱性を示さなくなる.IRE1αは複合体を形成し,自己リン酸化によって活性化され,mRNAやmiRNAを分解するRIDD(regulated IRE1-dependent decay)を起こす40).IRE1αの活性化は抗アポトーシス作用を持つmiRNAを分解することにより,細胞死を亢進させる41).MITOL欠損細胞では,IRE1αの複合体形成が促進し,IRE1αの活性化による細胞死が亢進する.MITOLはミトコンドリア外膜に局在し,IRE1αは小胞体膜に局在するが,ミトコンドリアと小胞体の接触部位であるMERCsに両者とも局在が認められる.MERCsにおいてMITOLはIRE1αの481番目のリシンにK63型のポリユビキチン鎖を付加する.IRE1α K481R変異体を細胞に発現させると,RIDDの亢進による細胞死が増加することから,MITOLによるIRE1αのユビキチン化修飾は,IRE1αの過剰な活性を防いでいる(図7).さらにMITOLノックアウトマウスを用いた解析においても,tunicamycin刺激によるIRE1αの過剰な活性化や細胞死の亢進が認められた42).このように,MITOLはミトコンドリア外膜やミトコンドリアに集積するタンパク質だけでなく,MERCsに局在する小胞体膜タンパク質に対しても,ユビキチン化修飾によりその活性化を制御することが明らかとなった.
アルツハイマー病は記憶や思考などに障害が起きる進行性の神経変性疾患であり,認知症の原因の一つである.認知症の患者数も年々増加しており,その原因の多くを占めるアルツハイマー病の詳細な病態解明や治療戦略の確立が求められている.アルツハイマー病発症初期の所見としてミトコンドリア機能障害が認められており43),アミロイドβの蓄積によってミトコンドリア機能障害が引き起こされる44).一方で,ミトコンドリアに異常タンパク質が蓄積した際に惹起されるミトコンドリアストレス応答をアルツハイマー病モデルマウスで誘導すると,アミロイドβの凝集の抑制やマウスの認知機能の改善が認められることから,ミトコンドリアストレス応答がアミロイドβ凝集の抑制にも寄与することが示唆されている45).また,アルツハイマー病の患者脳においてMERCsの増加が認められている46).小胞体からミトコンドリアへのMERCsを介した過剰なカルシウムの流入が細胞死を引き起こすことから,MERCsの増加はアルツハイマー病の一因となることが提唱されており47),ミトコンドリアやMERCsとアルツハイマー病との深い関連が示唆されている.
データベースAlzbaseでは,アルツハイマー病患者においてMITOLの発現の顕著な減少が報告されており48),MITOLとアルツハイマー病の関連が示唆された.我々は,アルツハイマー病におけるMITOLの役割の解明のために,アルツハイマー病モデルマウス(ADマウス;家族性アルツハイマー病関連遺伝子変異であるヒトAPPのSwedish変異とヒトプレセニリン1のexon 9欠損変異を導入したモデルマウス)と大脳皮質・海馬・嗅球特異的なMITOLノックアウトマウスを交配させ,MITOL欠損ADマウスを作出し,解析を行った.MITOL欠損ADマウスでは,内部構造が異常なミトコンドリアの増加などのミトコンドリア障害や,記憶力の低下が観察され,アルツハイマー病症状の増悪が認められ,MITOLがアルツハイマー病の発症を抑える働きを担うことが示唆された.MITOLの欠損はアミロイドβの産生量に大きな影響は与えないが,アミロイドβのオリゴマー形成を促進させる凝集性の高いプラークを形成させることが明らかとなった49).
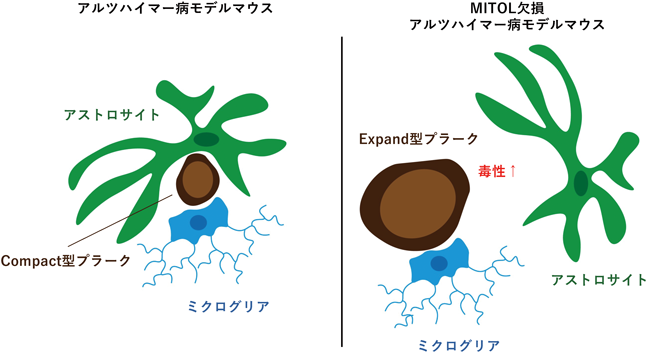
アミロイドβは高度に凝集するとβシート構造に富む線維構造を形成する.アミロイドβのプラーク内に形成された線維構造はアミロイドβプラークコアとして知られており,アミロイドβプラークコアはβシート構造特異的に結合するThioflavin S(ThS)によって染色できる50).MITOL欠損ADマウス脳においてThS陽性のアミロイドβプラークコアを持つプラークの形態に変化が認められた.アミロイドβプラークは形態学的にDiffuse型,Dense-core型,Compact型に分類される50).Diffuse型プラークは,プラーク形成の初期に観察され,ThS陽性アミロイド線維構造が認められない斑状の形態である.Dense-core型プラークは,プラークの中心部にThS陽性アミロイド線維構造のコアを有し,コアの周囲にアミロイドβ領域を持つ形態を指す.Compact型プラークは,アミロイドβ領域が圧縮され,高密度なアミロイド線維構造を持つプラークである51).病理所見からThS陽性アミロイド線維構造を持つプラークの周囲では神経細胞死が認められており,Dense-core型プラークは毒性を示す形態であり,Compact型プラークは弱毒化された形態と考えられる52, 53).MITOL欠損ADマウスでは,上記の定義のCompact型プラークの増加が認められたが,プラークサイズが大きく,周囲に神経変性が認められるプラークが高頻度で認められた.従来の毒性が低いとされるCompact型プラークとは異なる形質を示すプラークがMITOL欠損ADマウスで増加したと考えられる.そこで,ThS陽性のアミロイドβコアがプラーク全体の48%以上を占め,プラーク全体のサイズが40 mm2以上のサイズを持つアミロイドβプラークをExpand型プラークと分類した.新たな分類で比較定量すると,MITOL欠損ADマウスでは顕著に神経毒性の高いExpand型プラークが増加していた.MITOL欠損により増加したExpand型プラークが,アミロイドβのオリゴマー形成の誘導,細胞毒性の増加を引き起こした結果,MITOL欠損ADマウスのアルツハイマー症状が増悪したと考えられる54).
ミクログリアはアミロイドβプラークの周囲を取り囲み,グリアネットと呼ばれる構造体を形成することで,アミロイドβプラークの毒性を抑える55).大脳皮質・海馬・嗅球特異的MITOL欠損マウスでは,ミクログリアの異常な活性化が認められている15).そこでMITOL欠損によって,アミロイドβプラークに対するミクログリア応答に異常が生じ,正常にグリアネット形成が行われない可能性を検証したが,大脳皮質におけるミクログリア量やExpand型プラーク周囲へのミクログリアの集積量に大きな差は認められなかった.アストロサイトはミクログリアと同様にアミロイドβプラーク周辺を取り囲む構造を形成し,ThS陽性線維領域の拡大を抑制する56, 57).また,アストロサイトはプラークへの集積後にアミロイドβ分解酵素プリリジンの発現を介してアミロイドβ分解を行うことが知られている52).大脳皮質・海馬・嗅球特異的MITOL欠損マウスでは,アストロサイトの異常な活性化も認められている15).そこでMITOL欠損によって,アミロイドβプラークに対するアストロサイト応答に異常が生じ,アストロサイト集積が正常に行われない可能性を検証した.その結果,MITOL欠損ADマウスでは大脳皮質における活性化アストロサイト量の増加が認められた.一方で,アストロサイトのExpand型プラークへの集積が有意に減少していた.MITOL欠損によってアストロサイトの形質に変化が起きたためにプラークの毒性を回避できずに,毒性の高いExpand型プラークが増加し,アルツハイマー病の症状の悪化の原因の一つとなったと考えられる(図8).加齢に伴うミトコンドリア機能低下がアストロサイトのプラークの毒性回避作用を低下させ,Expand型プラークの増加を引き起こし,アルツハイマー病の発症や増悪の原因の一つとなる可能性が示唆された.MITOLがアストロサイトの形質変化に寄与する可能性は高いが,詳細なメカニズムの解明はされていない.今後,アストロサイトの形質変化におけるMITOLの役割の解明およびアルツハイマー病とのさらなる関連解明が期待される.
MITOL全身ノックアウトマウスは胎生致死である58).我々は生体内におけるMITOLの機能解明を行うために,部位特異的なMITOLノックアウトマウスを作出した.MITOLの2番目のエクソンの両端にLoxP配列を導入したマウスを作出し,部位特異的にCreリコンビナーゼを発現するマウスと交配させることにより,中枢神経系特異的(Nestin-promoter-Cre),大脳皮質・海馬・嗅球特異的(Emx1-promoter-Cre),表皮特異的(K14-promoter-Cre)などのMITOLノックアウトマウスを作出した.MITOL全身ノックアウトマウスが胎生致死であることやMITOLが胚性幹細胞の多分化能の維持に関与するとの知見から,中枢神経系特異的なMITOLノックアウトマウスも胎生致死となると予想していたが,中枢神経系特異的なMITOLノックアウトマウスは正常に生まれ,繁殖可能であり,1年以上の生存が確認された59).しかしながら,中枢神経系特異的なMITOLノックアウトマウスは雌雄ともに顕著な体重低下などの成長不全が観察された.体の成長を促す成長ホルモンは脳下垂体の前葉から分泌される.中枢神経系特異的なMITOLノックアウトマウスでは,脳下垂体の萎縮や成長ホルモンの分泌不全が認められた.pituitary transcription factor 1(Pit1)は脳下垂体特異的に発現する転写因子であり,成長ホルモンやプロラクチンの遺伝子発現を制御するだけでなく,成長ホルモン分泌細胞の分化や増殖にも重要な遺伝子である.MITOLを欠損した脳下垂体の生後0日,56日の両時期でPit1の発現減少が認められたことから,Pit1の発現減少が中枢神経特異的なMITOLノックアウトマウスの成長不全の原因の一因であることが明らかとなった59).Pit1はhomeobox protein prophet of Pit1(Prop1)によって発現が制御されているが60),MITOLを欠損した脳下垂体ではProp1のmRNAレベルに変化が認められなかった.MITOLの欠損によるPit1の発現低下の分子メカニズムとして,Prop1の活性低下が考えられる.Prop1とβ-cateninが結合することによりPit1の遺伝子発現を制御することや61),Mfn2がβ-cateninと結合し,β-cateninの核内移行を抑制することが報告されている62).MITOLはMfn2をユビキチン化によって制御することから,MITOLがMfn2/β-cateninを介してPit1の発現を制御する可能性が考えられる.今後,詳細な解析が期待される.
胚性幹細胞は分化多能性を保ちつつ,増殖可能な細胞である.Klf4は胚性幹細胞の多能性の維持に関わる転写因子である.マウス胚性幹細胞であるR1細胞にレトロウイルスを用いて胚性幹細胞の多能性の維持に関与する因子を探索した結果,胚性幹細胞の多能性の維持を担う因子としてMITOLが同定されている63).レチノイン酸を用いてR1細胞を分化させた際,分化の経過とともにMITOLの発現が顕著に減少する.さらにマウス胎仔線維芽細胞(MEFs)をiPS細胞にリプログラミングするとMITOLの発現量は増加することから,MITOLは多能性幹細胞に多く発現する.興味深いことに,iPS細胞を作成する際に導入する四つの遺伝子(Oct4, Sox2, Klf4, c-Myc)のうちの一つであるKlf4によってMITOLの発現が制御されており,MITOLがKlf4による細胞の多能性の獲得に一部貢献している.MITOLはprotein kinase A(PKA)の活性抑制に働くPrkar1aにK63型のポリユビキチン化を付加し,Prkar1aとPKAの結合を解離させる.PKAはERKの活性化を抑制に働くことから,MITOLの欠失により,PKAは抑制され,ERKが活性化する.MITOLはPrkar1aを介してERKシグナリングを制御することにより細胞の多能性に関与する63).マウス脳においても胎生期にMITOLの発現量が多く生後8週齢では劇的に減少することから15),MITOLは幹細胞に多く,細胞分化に関わる可能性が示唆される.
ミトコンドリアはRNAウイルスに対して細胞内の自然免疫応答に関与する.ミトコンドリアに局在するmitochondrial antiviral signalling(MAVS,別名VISA)は,細胞内のウイルスのRNAを認識するセンサーであるretinoic acid-inducible gene I(RIG-I),melanoma differentiation-associated gene 5(MDA-5)やLGP2と相互作用し,NFκBの活性化など下流にシグナルを伝え,1型interferon(IFN)および炎症性サイトカインの産生を促し,抗ウイルス防御機構をもたらす.しかしながら,過剰な炎症反応の亢進は細胞にとって有害であることから,MAVSを介した炎症反応の調節は必要である.MITOLはMAVSにK48型のポリユビキチン鎖を付加し,プロテアソームによる分解を促進することにより,MAVSの発現量を調節し,過剰な炎症反応の亢進を防ぐ機能を有する58).MAVSの過剰な活性化は疾患との関連も報告されている.全身性エリテマトーデス(systemic lupus erythematosus:SLE)は日本全国に6万~10万人ほどの患者がいる指定難病であり,全身のさまざまな臓器に炎症や障害を起こす自己免疫疾患である.SLE患者では,IFNやIFNによって誘導される遺伝子群の発現量の増加が認められている.MAVSはIFNの発現制御に関わることから,MAVS遺伝子の遺伝子多型がSLEの発症のしやすさや症状に影響を与える可能性が示唆されている64).SLE患者由来の細胞から抽出したサンプルを半変性洗剤アガロースゲル電気泳動(SDD-AGE)を用いて解析すると,MAVSの凝集およびそれに伴うIFNの増加が認められており,SLEの症状悪化にMAVSの凝集が関与することが示唆されている65).MITOLはユビキチン–プロテアソーム系を介してMAVSの分解を促進する58).MAVSの凝集が認められるSLE患者の細胞では,MITOLの発現量の減少が認められていることから,MITOLの欠失がMAVSの凝集,IFNの活性化を引き起こし,SLEの症状を増悪させるメカニズムの一つと考えられている65).MAVSの負の制御系として,PKAによるリン酸化修飾がある.MITOLはリン酸化修飾されたMAVSのユビキチン–プロテアソームによる分解を促進することにより,MAVSの過剰な活性化を抑制する66).MITOLはミトコンドリア外膜上においてMAVSを介して自然免疫応答を制御する役割を担っている.
マイトファジーは障害を受けたミトコンドリアや不要なミトコンドリアを特異的に分解するオートファジーであり,ミトコンドリアの品質管理に重要な役割を担っている.FUNDC1はマイトファジー受容体の一つであり,低酸素ストレス下においてPGAM5によって脱リン酸化されることによりLC1との結合が増加し,障害ミトコンドリアのマイトファジーによる分解を促進する67).MITOLはFUNDC1と結合し,ユビキチン–プロテアソーム系によってその分解を促進する.MITOLの欠失はFUNDC1を蓄積させ,低酸素ストレスにより誘導されるマイトファジーを増加させる68).低酸素の条件下で,MITOLはFUNDC1と結合し,分解を促進することでマイトファジーからミトコンドリアを守っているが,持続的または重度の低酸素の条件下では,FUNDC1は脱リン酸化され,安定化し,マイトファジーを引き起こす.MITOLは細胞内ストレスの度合いに応じたマイトファジーの調節因子といえる.
MITOLはParkin依存的なマイトファジーとの関連についても報告されている.ミトコンドリアが障害を受け,ミトコンドリアの膜電位が消失するとPINK1がミトコンドリアの外膜で安定化され,PINK1がユビキチンをリン酸化する.リン酸化ユビキチンはParkinを活性化し,損傷ミトコンドリア上にポリユビキチン鎖が形成され,オートファジーによって,損傷ミトコンドリアが分解される69).MITOLはcarbonyl cyanide m-chlorophenylhydrazone(CCCP)により脱分極したミトコンドリアに集積したParkin特異的に結合し,Parkinの220番目のリシンにK48型のポリユビキチン鎖を付加し,Parkinの分解を促進する.MITOL欠損細胞ではParkin依存的なマイトファジーが亢進するのに対し,MITOLを過剰発現した細胞ではParkin依存的なマイトファジーが抑制される.ミトコンドリアの損傷が軽度のとき,MITOLはParkin依存的なマイトファジーが過剰に働くのを抑える役割を担うことが示唆された.マイトファジーにおいて,MITOLとParkinのどちらが優位に働くか,その詳細な調節機構に関してはいまだ不明である.興味深いことに,Parkin依存的なマイトファジーの際,Tom20などのミトコンドリア外膜タンパク質と比べて,MITOLの分解速度は非常に遅い.CCCP処理24時間後のMITOLの局在がCalnexin陽性の小胞体と一致することから,マイトファジーによってミトコンドリアが分解されると,MITOLが小胞体に移行することが明らかとなった.FK506-binding protein 38(FKBP38)は主にミトコンドリアに局在し,抗アポトーシス機能を有する.マイトファジーの際,FKBP38はミトコンドリアから小胞体へ移行し,マイトファジーからの分解を逃れ,アポトーシスを抑制することが報告されている70).MITOLもFKBP38と同様に,ミトコンドリアから小胞体に移行したと考えられる.さらに,MITOL欠損細胞では,FKBP38の分解が促進され,細胞死が亢進することが認められた.MITOLはマイトファジーの際に小胞体に移行し,ParkinによるFKBP38の分解を抑制し,細胞死を抑制する役割を担うことが明らかとなった.ミトコンドリア外膜貫通型のタンパク質であるMITOLが小胞体へ移行する詳細なメカニズムは不明であるが,MITOLは小胞体移行後にもParkinの過剰な活性化による細胞死を防ぐ役割を担う.Parkinによるミトコンドリアの除去機構と小胞体における抗アポトーシス分子の分解機構の二つの段階でMITOLはParkinを調節していると考えられる.
一方で,MITOLがParkinのミトコンドリアへの局在を助けているとの報告もある71).MITOLを欠失すると,CCCP処理後のParkinのミトコンドリアへのリクルートが遅くなる.MITOLによってユビキチン化されたミトコンドリア外膜タンパク質のユビキチン鎖をPINK1がリン酸化し,リン酸化ユビキチンを認識したParkinがミトコンドリアへ集積する71).さらに,マイトファジーによりミトコンドリアが分解された際にMITOLがミトコンドリアからペルオキシソームに移行することも報告されている72).ペルオキシソーム移行後のMITOLの役割は明らかとされていないが,MITOLのC末端を欠損させた変異体MITOLΔC8を発現させると,ペルオキシソームの膨潤が確認されており,MITOLがペルオキシソームにおいて何らかの役割を担うことが示唆されている.近年,マイトファジー条件下ではなく,定常状態においてもMITOLはペルオキシソームに局在することが報告されている73).MITOLはPEX3, PEX19, peroxisome membrane protein 70(PMP70)と結合する.飢餓刺激によって,PMP70のユビキチン化がペキソファジーを制御することが報告されている74, 75).mTOR阻害剤であるTorin1によって誘導されるペキソファジー条件下において,MITOLはPMP70をユビキチン化修飾し,ペキソファジーを誘導する.MITOL欠損細胞では,Torin1に誘導されるペキソファジーの抑制が認められる73).Parkin依存的なマイトファジー条件下においては,ペルオキシソームの数に大きな違いが認められていないことから,MITOLはペキソファジー以外の役割を担っていると推察される.今後,内在性Parkinが発現する細胞にて,Parkin依存的なマイトファジーにおけるMITOLの役割の解明が期待される.また,ミトコンドリアの分解後に,MITOLがどこに局在し,機能するのか今後の研究が期待される.
ミトコンドリアの外膜では多くの細胞内イベントが起きている.ミトコンドリア外膜に局在するMITOLはミトコンドリア外膜上で基質をユビキチン化し,制御する役割を担っており,ミトコンドリアダイナミクス,MERCsの形成制御,免疫応答,マイトファジーとさまざまな機構との関連が示されている.MITOLは臓器や細胞種の違いによって異なる役割を持つことが示唆されているが,生体内におけるMITOLの機能解析に関する研究報告は少ない.今後,マウスなどの個体やオルガノイドを用いた解析により新たな知見が得られ,臓器や細胞種ごとのMITOLの役割やMITOLの制御機構が解明されることが期待される.
謝辞Acknowledgments
これまでにご指導いただいた先生方ならびに研究にご協力いただいたすべての方々に深く御礼申し上げます.
引用文献References
1) Gomes, L.C., Di Benedetto, G., & Scorrano, L. (2011) During autophagy mitochondria elongate, are spared from degradation and sustain cell viability. Nat. Cell Biol., 13, 589–598.
2) Rambold, A.S., Kostelecky, B., Elia, N., & Lippincott-Schwartz, J. (2011) Tubular network formation protects mitochondria from autophagosomal degradation during nutrient starvation. Proc. Natl. Acad. Sci. USA, 108, 10190–10195.
3) Tilokani, L., Nagashima, S., Paupe, V., & Prudent, J. (2018) Mitochondrial dynamics: overview of molecular mechanisms. Essays Biochem., 62, 341–360.
4) Liu, Y., Ma, X., Fujioka, H., Liu, J., Chen, S., & Zhu, X. (2019) DJ-1 regulates the integrity and function of ER-mitochondria association through interaction with IP3R3-Grp75-VDAC1. Proc. Natl. Acad. Sci. USA, 116, 25322–25328.
5) de Brito, O.M. & Scorrano, L. (2008) Mitofusin 2 tethers endoplasmic reticulum to mitochondria. Nature, 456, 605–610.
6) De Vos, K.J., Mórotz, G.M., Stoica, R., Tudor, E.L., Lau, K.F., Ackerley, S., Warley, A., Shaw, C.E., & Miller, C.C. (2012) VAPB interacts with the mitochondrial protein PTPIP51 to regulate calcium homeostasis. Hum. Mol. Genet., 21, 1299–1311.
7) Bauer, T.M. & Murphy, E. (2020) Role of mitochondrial calcium and the permeability transition pore in regulating cell death. Circ. Res., 126, 280–293.
8) Lemasters, J.J., Theruvath, T.P., Zhong, Z., & Nieminen, A.L. (2009) Mitochondrial calcium and the permeability transition in cell death. Biochim. Biophys. Acta, 1787, 1395–1401.
9) Bonora, M., Patergnani, S., Ramaccini, D., Morciano, G., Pedriali, G., Kahsay, A.E., Bouhamida, E., Giorgi, C., Wieckowski, M.R., & Pinton, P. (2020) Physiopathology of the permeability transition pore: Molecular mechanisms in human pathology. Biomolecules, 10, 998.
10) Hirabayashi, Y., Kwon, S.K., Paek, H., Pernice, W.M., Paul, M.A., Lee, J., Erfani, P., Raczkowski, A., Petrey, D.S., Pon, L.A., et al. (2017) ER-mitochondria tethering by PDZD8 regulates Ca(2+) dynamics in mammalian neurons. Science, 358, 623–630.
11) Iwasawa, R., Mahul-Mellier, A.L., Datler, C., Pazarentzos, E., & Grimm, S. (2011) Fis1 and Bap31 bridge the mitochondria-ER interface to establish a platform for apoptosis induction. EMBO J., 30, 556–568.
12) Bartee, E., Mansouri, M., Hovey Nerenberg, B.T., Gouveia, K., & Früh, K. (2004) Downregulation of major histocompatibility complex class I by human ubiquitin ligases related to viral immune evasion proteins. J. Virol., 78, 1109–1120.
13) Yonashiro, R., Ishido, S., Kyo, S., Fukuda, T., Goto, E., Matsuki, Y., Ohmura-Hoshino, M., Sada, K., Hotta, H., Yamamura, H., et al. (2006) A novel mitochondrial ubiquitin ligase plays a critical role in mitochondrial dynamics. EMBO J., 25, 3618–3626.
14) Kim, S.H., Park, Y.Y., Yoo, Y.S., & Cho, H. (2016) Self-clearance mechanism of mitochondrial E3 ligase MARCH5 contributes to mitochondria quality control. FEBS J., 283, 294–304.
15) Nagashima, S., Takeda, K., Ohno, N., Ishido, S., Aoki, M., Saitoh, Y., Takada, T., Tokuyama, T., Sugiura, A., Fukuda, T., et al. (2019) MITOL deletion in the brain impairs mitochondrial structure and ER tethering leading to oxidative stress. Life Sci. Alliance, 2, e201900308.
16) Pernas, L. & Scorrano, L. (2016) Mito-Morphosis: Mitochondrial fusion, fission, and cristae remodeling as key mediators of cellular function. Annu. Rev. Physiol., 78, 505–531.
17) Nagashima, S., Tábara, L.C., Tilokani, L., Paupe, V., Anand, H., Pogson, J.H., Zunino, R., McBride, H.M., & Prudent, J. (2020) Golgi-derived PI(4)P-containing vesicles drive late steps of mitochondrial division. Science, 367, 1366–1371.
18) Wong, Y.C., Ysselstein, D., & Krainc, D. (2018) Mitochondria-lysosome contacts regulate mitochondrial fission via RAB7 GTP hydrolysis. Nature, 554, 382–386.
19) Friedman, J.R., Lackner, L.L., West, M., DiBenedetto, J.R., Nunnari, J., & Voeltz, G.K. (2011) ER tubules mark sites of mitochondrial division. Science, 334, 358–362.
20) Karbowski, M., Neutzner, A., & Youle, R.J. (2007) The mitochondrial E3 ubiquitin ligase MARCH5 is required for Drp1 dependent mitochondrial division. J. Cell Biol., 178, 71–84.
21) Xu, S., Cherok, E., Das, S., Li, S., Roelofs, B.A., Ge, S.X., Polster, B.M., Boyman, L., Lederer, W.J., Wang, C., et al. (2016) Mitochondrial E3 ubiquitin ligase MARCH5 controls mitochondrial fission and cell sensitivity to stress-induced apoptosis through regulation of MiD49 protein. Mol. Biol. Cell, 27, 349–359.
22) Cherok, E., Xu, S., Li, S., Das, S., Meltzer, W.A., Zalzman, M., Wang, C., & Karbowski, M. (2017) Novel regulatory roles of Mff and Drp1 in E3 ubiquitin ligase MARCH5-dependent degradation of MiD49 and Mcl1 and control of mitochondrial dynamics. Mol. Biol. Cell, 28, 396–410.
23) Davalli, P., Mitic, T., Caporali, A., Lauriola, A., & D’Arca, D. (2016) ROS, cell senescence, and novel molecular mechanisms in aging and age-Related diseases. Oxid. Med. Cell. Longev., 2016, 3565127.
24) Yu, B., Ma, J., Li, J., Wang, D., Wang, Z., & Wang, S. (2020) Mitochondrial phosphatase PGAM5 modulates cellular senescence by regulating mitochondrial dynamics. Nat. Commun., 11, 2549.
25) Yoon, Y.S., Yoon, D.S., Lim, I.K., Yoon, S.H., Chung, H.Y., Rojo, M., Malka, F., Jou, M.J., Martinou, J.C., & Yoon, G. (2006) Formation of elongated giant mitochondria in DFO-induced cellular senescence: involvement of enhanced fusion process through modulation of Fis1. J. Cell. Physiol., 209, 468–480.
26) Park, Y.Y., Lee, S., Karbowski, M., Neutzner, A., Youle, R.J., & Cho, H. (2010) Loss of MARCH5 mitochondrial E3 ubiquitin ligase induces cellular senescence through dynamin-related protein 1 and mitofusin 1. J. Cell Sci., 123, 619–626.
27) Taguchi, N., Ishihara, N., Jofuku, A., Oka, T., & Mihara, K. (2007) Mitotic phosphorylation of dynamin-related GTPase Drp1 participates in mitochondrial fission. J. Biol. Chem., 282, 11521–11529.
28) Park, Y.Y. & Cho, H. (2012) Mitofusin 1 is degraded at G2/M phase through ubiquitylation by MARCH5. Cell Div., 7, 25.
29) Park, Y.Y., Nguyen, O.T., Kang, H., & Cho, H. (2014) MARCH5-mediated quality control on acetylated Mfn1 facilitates mitochondrial homeostasis and cell survival. Cell Death Dis., 5, e1172.
30) Naon, D., Zaninello, M., Giacomello, M., Varanita, T., Grespi, F., Lakshminaranayan, S., Serafini, A., Semenzato, M., Herkenne, S., Hernández-Alvarez, M.I., et al. (2016) Critical reappraisal confirms that Mitofusin 2 is an endoplasmic reticulum-mitochondria tether. Proc. Natl. Acad. Sci. USA, 113, 11249–11254.
31) Hernández-Alvarez, M.I., Sebastián, D., Vives, S., Ivanova, S., Bartoccioni, P., Kakimoto, P., Plana, N., Veiga, S.R., Hernández, V., Vasconcelos, N., et al. (2019) Deficient endoplasmic reticulum-Mitochondrial phosphatidylserine transfer causes liver disease. Cell, 177, 881–95.e17.
32) Nakamura, N., Kimura, Y., Tokuda, M., Honda, S., & Hirose, S. (2006) MARCH-V is a novel mitofusin 2- and Drp1-binding protein able to change mitochondrial morphology. EMBO Rep., 7, 1019–1022.
33) Sugiura, A., Nagashima, S., Tokuyama, T., Amo, T., Matsuki, Y., Ishido, S., Kudo, Y., McBride, H.M., Fukuda, T., Matsushita, N., et al. (2013) MITOL regulates endoplasmic reticulum-mitochondria contacts via Mitofusin2. Mol. Cell, 51, 20–34.
34) Gomez-Suaga, P., Paillusson, S., Stoica, R., Noble, W., Hanger, D.P., & Miller, C.C.J. (2017) The ER-mitochondria tethering complex VAPB-PTPIP51 regulates autophagy. Curr. Biol., 27, 371–385.
35) Yeo, H.K., Park, T.H., Kim, H.Y., Jang, H., Lee, J., Hwang, G.S., Ryu, S.E., Park, S.H., Song, H.K., Ban, H.S., et al. (2021) Phospholipid transfer function of PTPIP51 at mitochondria-associated ER membranes. EMBO Rep., 22, e51323.
36) Ito, N., Takahashi, T., Shiiba, I., Nagashima, S., Inatome, R., & Yanagi, S. (2021) MITOL regulates phosphatidic acid-binding activity of RMDN3/PTPIP51. J. Biochem., 171, 529–541.
37) Shore, G.C., Papa, F.R., & Oakes, S.A. (2011) Signaling cell death from the endoplasmic reticulum stress response. Curr. Opin. Cell Biol., 23, 143–149.
38) Tabas, I. & Ron, D. (2011) Integrating the mechanisms of apoptosis induced by endoplasmic reticulum stress. Nat. Cell Biol., 13, 184–190.
39) Yoshida, H. (2007) ER stress and diseases. FEBS J., 274, 630–658.
40) Hollien, J., Lin, J.H., Li, H., Stevens, N., Walter, P., & Weissman, J.S. (2009) Regulated Ire1-dependent decay of messenger RNAs in mammalian cells. J. Cell Biol., 186, 323–331.
41) Lerner, A.G., Upton, J.P., Praveen, P.V., Ghosh, R., Nakagawa, Y., Igbaria, A., Shen, S., Nguyen, V., Backes, B.J., Heiman, M., et al. (2012) IRE1α induces thioredoxin-interacting protein to activate the NLRP3 inflammasome and promote programmed cell death under irremediable ER stress. Cell Metab., 16, 250–264.
42) Takeda, K., Nagashima, S., Shiiba, I., Uda, A., Tokuyama, T., Ito, N., Fukuda, T., Matsushita, N., Ishido, S., Iwawaki, T., et al. (2019) MITOL prevents ER stress-induced apoptosis by IRE1α ubiquitylation at ER-mitochondria contact sites. EMBO J., 38, e100999.
43) Selfridge, J.E., e, L., Lu, J., & Swerdlow, R.H. (2013) Role of mitochondrial homeostasis and dynamics in Alzheimer’s disease. Neurobiol. Dis., 51, 3–12.
44) Chen, J.X. & Yan, S.D. (2007) Amyloid-beta-induced mitochondrial dysfunction. J. Alzheimers Dis., 12, 177–184.
45) Sorrentino, V., Romani, M., Mouchiroud, L., Beck, J.S., Zhang, H., D’Amico, D., Moullan, N., Potenza, F., Schmid, A.W., Rietsch, S., et al. (2017) Enhancing mitochondrial proteostasis reduces amyloid-β proteotoxicity. Nature, 552, 187–193.
46) Hedskog, L., Pinho, C.M., Filadi, R., Rönnbäck, A., Hertwig, L., Wiehager, B., Larssen, P., Gellhaar, S., Sandebring, A., Westerlund, M., et al. (2013) Modulation of the endoplasmic reticulum-mitochondria interface in Alzheimer’s disease and related models. Proc. Natl. Acad. Sci. USA, 110, 7916–7921.
47) Area-Gomez, E., Del Carmen Lara Castillo, M., Tambini, M.D., Guardia-Laguarta, C., de Groof, A.J., Madra, M., Ikenouchi, J., Umeda, M., Bird, T.D., Sturley, S.L., et al. (2012) Upregulated function of mitochondria-associated ER membranes in Alzheimer disease. EMBO J., 31, 4106–4123.
48) Bai, Z., Han, G., Xie, B., Wang, J., Song, F., Peng, X., & Lei, H. (2016) AlzBase: an Integrative Database for Gene Dysregulation in Alzheimer’s Disease. Mol. Neurobiol., 53, 310–319.
49) Takeda, K., Uda, A., Mitsubori, M., Nagashima, S., Iwasaki, H., Ito, N., Shiiba, I., Ishido, S., Matsuoka, M., Inatome, R., et al. (2021) Mitochondrial ubiquitin ligase alleviates Alzheimer’s disease pathology via blocking the toxic amyloid-β oligomer generation. Commun. Biol., 4, 192.
50) Dickson, T.C. & Vickers, J.C. (2001) The morphological phenotype of beta-amyloid plaques and associated neuritic changes in Alzheimer’s disease. Neuroscience, 105, 99–107.
51) Bussière, T., Bard, F., Barbour, R., Grajeda, H., Guido, T., Khan, K., Schenk, D., Games, D., Seubert, P., & Buttini, M. (2004) Morphological characterization of Thioflavin-S-positive amyloid plaques in transgenic Alzheimer mice and effect of passive Abeta immunotherapy on their clearance. Am. J. Pathol., 165, 987–995.
52) Michno, W., Wehrli, P., Meier, S.R., Sehlin, D., Syvänen, S., Zetterberg, H., Blennow, K., & Hanrieder, J. (2020) Chemical imaging of evolving amyloid plaque pathology and associated Aβ peptide aggregation in a transgenic mouse model of Alzheimer’s disease. J. Neurochem., 152, 602–616.
53) Condello, C., Schain, A., & Grutzendler, J. (2011) Multicolor time-stamp reveals the dynamics and toxicity of amyloid deposition. Sci. Rep., 1, 19.
54) Mitsubori, M., Takeda, K., Nagashima, S., Ishido, S., Matsuoka, M., Inatome, R., & Yanagi, S. (2021) Identification of highest neurotoxic amyloid-β plaque type showing reduced contact with astrocytes. Biochem. Biophys. Res. Commun., 549, 67–74.
55) Condello, C., Yuan, P., Schain, A., & Grutzendler, J. (2015) Microglia constitute a barrier that prevents neurotoxic protofibrillar Aβ42 hotspots around plaques. Nat. Commun., 6, 6176.
56) Bouvier, D.S., Jones, E.V., Quesseveur, G., Davoli, M.A., A Ferreira, T., Quirion, R., Mechawar, N., & Murai, K.K. (2016) High resolution dissection of reactive glial nets in alzheimer’s disease. Sci. Rep., 6, 24544.
57) Smith, A.J., Duan, T., & Verkman, A.S. (2019) Aquaporin-4 reduces neuropathology in a mouse model of Alzheimer’s disease by remodeling peri-plaque astrocyte structure. Acta Neuropathol. Commun., 7, 74.
58) Yoo, Y.S., Park, Y.Y., Kim, J.H., Cho, H., Kim, S.H., Lee, H.S., Kim, T.H., Sun Kim, Y., Lee, Y., Kim, C.J., et al. (2015) The mitochondrial ubiquitin ligase MARCH5 resolves MAVS aggregates during antiviral signalling. Nat. Commun., 6, 7910.
59) Matsuno, K., Nagashima, S., Shiiba, I., Taniwaka, K., Takeda, K., Tokuyama, T., Ito, N., Matsushita, N., Fukuda, T., Ishido, S., et al. (2020) MITOL dysfunction causes dwarfism with anterior pituitary hypoplasia. J. Biochem., 168, 305–312.
60) Ikeshita, N., Kawagishi, M., Shibahara, H., Toda, K., Yamashita, T., Yamamoto, D., Sugiyama, Y., Iguchi, G., Iida, K., Takahashi, Y., et al. (2008) Identification and analysis of prophet of Pit-1-binding sites in human Pit-1 gene. Endocrinology, 149, 5491–5499.
61) Olson, L.E., Tollkuhn, J., Scafoglio, C., Krones, A., Zhang, J., Ohgi, K.A., Wu, W., Taketo, M.M., Kemler, R., Grosschedl, R., et al. (2006) Homeodomain-mediated beta-catenin-dependent switching events dictate cell-lineage determination. Cell, 125, 593–605.
62) Kim, Y.M., Krantz, S., Jambusaria, A., Toth, P.T., Moon, H.G., Gunarathna, I., Park, G.Y., & Rehman, J. (2021) Mitofusin-2 stabilizes adherens junctions and suppresses endothelial inflammation via modulation of β-catenin signaling. Nat. Commun., 12, 2736.
63) Gu, H., Li, Q., Huang, S., Lu, W., Cheng, F., Gao, P., Wang, C., Miao, L., Mei, Y., & Wu, M. (2015) Mitochondrial E3 ligase March5 maintains stemness of mouse ES cells via suppression of ERK signalling. Nat. Commun., 6, 7112.
64) Liu, X., Jiao, Y., Wen, X., Wang, L., Ma, C., Gao, X., Chen, Z.J., & Zhao, Y. (2011) Possible association of VISA gene polymorphisms with susceptibility to systemic lupus erythematosus in Chinese population. Mol. Biol. Rep., 38, 4583–4588.
65) Shao, W.H., Shu, D.H., Zhen, Y., Hilliard, B., Priest, S.O., Cesaroni, M., Ting, J.P., & Cohen, P.L. (2016) Prion-like aggregation of mitochondrial antiviral signaling protein in lupus patients is associated with increased levels of type I interferon. Arthritis Rheumatol., 68, 2697–2707.
66) Yan, B.R., Zhou, L., Hu, M.M., Li, M., Lin, H., Yang, Y., Wang, Y.Y., & Shu, H.B. (2017) PKACs attenuate innate antiviral response by phosphorylating VISA and priming it for MARCH5-mediated degradation. PLoS Pathog., 13, e1006648.
67) Liu, L., Feng, D., Chen, G., Chen, M., Zheng, Q., Song, P., Ma, Q., Zhu, C., Wang, R., Qi, W., et al. (2012) Mitochondrial outer-membrane protein FUNDC1 mediates hypoxia-induced mitophagy in mammalian cells. Nat. Cell Biol., 14, 177–185.
68) Chen, Z., Siraj, S., Liu, L., & Chen, Q. (2017) MARCH5-FUNDC1 axis fine-tunes hypoxia-induced mitophagy. Autophagy, 13, 1244–1245.
69) Onishi, M., Yamano, K., Sato, M., Matsuda, N., & Okamoto, K. (2021) Molecular mechanisms and physiological functions of mitophagy. EMBO J., 40, e104705.
70) Saita, S., Shirane, M., & Nakayama, K.I. (2013) Selective escape of proteins from the mitochondria during mitophagy. Nat. Commun., 4, 1410.
71) Koyano, F., Yamano, K., Kosako, H., Tanaka, K., & Matsuda, N. (2019) Parkin recruitment to impaired mitochondria for nonselective ubiquitylation is facilitated by MITOL. J. Biol. Chem., 294, 10300–10314.
72) Koyano, F., Yamano, K., Kosako, H., Kimura, Y., Kimura, M., Fujiki, Y., Tanaka, K., & Matsuda, N. (2019) Parkin-mediated ubiquitylation redistributes MITOL/March5 from mitochondria to peroxisomes. EMBO Rep., 20, e47728.
73) Zheng, J., Chen, X., Liu, Q., Zhong, G., & Zhuang, M. (2022) Ubiquitin ligase MARCH5 localizes to peroxisomes to regulate pexophagy. J. Cell Biol., 221, e202103156.
74) Kim, P.K., Hailey, D.W., Mullen, R.T., & Lippincott-Schwartz, J. (2008) Ubiquitin signals autophagic degradation of cytosolic proteins and peroxisomes. Proc. Natl. Acad. Sci. USA, 105, 20567–20574.
75) Sargent, G., van Zutphen, T., Shatseva, T., Zhang, L., Di Giovanni, V., Bandsma, R., & Kim, P.K. (2016) PEX2 is the E3 ubiquitin ligase required for pexophagy during starvation. J. Cell Biol., 214, 677–690.
著者紹介Author Profile
 長島 駿(ながしま しゅん)
長島 駿(ながしま しゅん)東京薬科大学生命科学部再生医科学研究室助教.博士(生命科学).
略歴1985年東京都に生る.2007年東京薬科大学生命科学部卒業.12年同大学院博士課程修了.同年東京薬科大学助教,17年Cambridge大学研究員を経て,19年より現職.
研究テーマと抱負生体内におけるミトコンドリアを中心としたオルガネラコンタクトの役割に興味を持ち研究を進めている.現在はヒト初期発生や各臓器におけるミトコンドリアの役割の解明を目指している.
ウェブサイトhttps://www.toyaku.ac.jp/lifescience/labo/lifescience03/regenerativemedicine.html
趣味サッカー,フットサル.