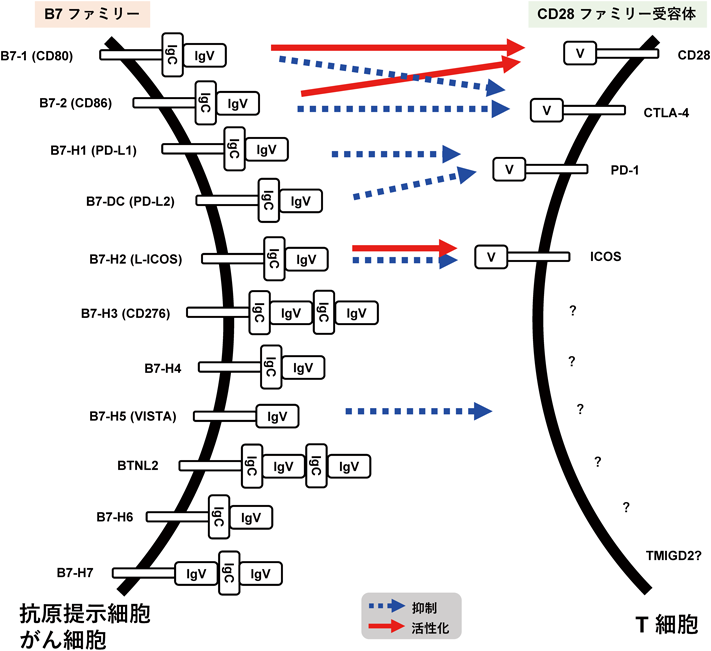
生体内においてがん細胞は免疫チェックポイント分子を介した免疫寛容を引き起こすことで,免疫系による排除を回避する.T細胞上に発現するCD28ファミリー受容体(CD28, CTLA-4, PD-1やICOSなど)と抗原提示細胞やがん細胞上に発現するB7ファミリーリガンドの結合はT細胞に対し負のシグナルを伝達し,その結果としてT細胞の活性化や増殖が抑制される.現時点では,B7-1(CD80)をはじめとし,B7-2, B7-H1(PD-L1),B7-DC(PD-L2),B7-H2, B7-H3, B7-H4, B7-H5, BTNL2, B7-H6, B7-H7の11個のB7ファミリーリガンドが同定されている(図1).
上述のように,PD-L1やPD-L2はPD-1のリガンドとして働く.PD-L1による免疫寛容の仕組みとしては,まずPD-L1がT細胞上のPD-1と相互作用することで,PD-1の細胞内ドメインに存在するチロシン残基のリン酸化を引き起こす.そして,これによりT細胞受容体(TCR)を介したシグナル伝達分子とその関連分子のリン酸化レベルの低下がもたらされ,結果としてT細胞の活性化やサイトカイン産生が抑制される1).PD-L1は大腸がん,肝細胞がん,肺がん,胃がん,膵臓がん,卵巣がんなど,さまざまながん組織で発現の上昇が報告されている.また,子宮頚がんや卵巣がん,非小細胞肺がんにおいては,PD-L1の高発現と予後不良の相関が報告されている2).
PD-1以外のCD28ファミリー受容体であるCD28やCTLA-4に対しては,CD80(B7-1)やCD86(B7-2)が結合する.また,L-ICOS(B7-H2)はT細胞上のICOSに結合するリガンドであることが知られている.しかし,B7-H3, B7-H4, B7-H5, BTNL2, B7-H6, B7-H7等のB7ファミリーリガンドがどのCD28ファミリー受容体に結合するかはまだはっきりしていない.ほとんどのB7ファミリーリガンドは免疫グロブリン(Ig)の可変領域様(IgV-like)ドメインと定常領域様(IgC-like)ドメインの両方を有しているが,B7-H5(VISTA)がIgV-likeドメインだけを持っている.一方,CD28ファミリー受容体もIgV-likeドメインだけを持つ.このことから,VISTAはリガンドとしての特性に加え,受容体としての性質を併せ持つ可能性が想定されている.VISTAが受容体として機能するかについては現時点では不明であるが,骨髄由来の樹状細胞にVISTAを高発現させるとT細胞の増殖やサイトカイン産生が抑制されるという報告があるため,免疫寛容を引き起こすリガンドとしての機能は有していると考えられている3).
PD-1やPD-L1などの免疫チェックポイント分子に対する抗体薬(免疫チェックポイント阻害剤)は,PD-1とPD-L1の結合を物理的に阻害することができることから,PD-1/PD-L1によって引き起こされる免疫寛容を食い止めることができる.事実,PD-1やPD-L1に対する抗体薬は悪性黒色腫や腎細胞がん,非小細胞肺がんを対象とした臨床試験において有効性が示されている.近年ではさまざまな製薬企業からPD-1やPD-L1に対する抗体薬が開発され,アメリカ食品医薬品局(FDA)から承認されている4).しかし,PD-1やPD-L1に対する抗体薬の抗腫瘍効果は示されているものの,患者によっては奏功率が低いことも明らかとなっている.このことから,免疫チェックポイント阻害剤の奏功率を向上させるために,他の抗がん剤や分子標的薬を免疫チェックポイント阻害薬と併用する複合的がん免疫療法が期待され,欧米では多数の臨床試験がすすめられている.また,PD-L1の発現や機能を制御する低分子化合物は新たな分子標的薬として免疫チェックポイント阻害剤との併用療法に有用であると考えられている.中でもPD-L1の発現や機能を制御する翻訳後修飾に対する低分子化合物はその分子標的薬の候補になると期待されている.そこで,本稿ではPD-L1の発現や細胞内機能を制御する翻訳後修飾について概説する.
1)DNA損傷刺激
放射線やDNA傷害性抗がん剤(カンプトテシン,アドリアマイシン,エトポシドなど)の添加によるDNA損傷刺激は転写レベルでPD-L1の発現を上昇させる.興味深いことに,アドリアマイシンは細胞膜上のPD-L1タンパク質の発現量を低下させ,核内の発現量を上昇させることが報告されている5).DNA損傷応答はゲノムの安定性の維持に必須な分子機構である.損傷を受けたDNAの傷は,DNA損傷のセンサー分子であるataxia telangiectasia mutated(ATM)やataxia telangiectasia and Rad3 related protein(ATR)によって認識され,自己リン酸化によって活性化したATMやATRは基質であるcheckpoint kinase 1/2(CHK1/2)やp53をリン酸化することでDNA損傷シグナルを下流へと伝える.DNA二本鎖切断(DNA double-strand breaks:DSBs)や一本鎖切断(single-strand breaks:SSBs)による損傷箇所は相同組換え(homologous recombination:HR)もしくは非相同末端結合(non-homologous end-joining:NHEJ)によって修復される.poly(ADP-ribose)polymerases 1(PARP-1)は,DNA損傷によって生じた一本鎖切断端を認識してDNAに結合する.DNAに結合したPARPは活性化され,細胞内のニコチンアミドアデニンジヌクレオチド(NAD+)依存的にADPリボースをPARP自身やDNA損傷修復関連タンパク質[RCC1, DNA ligase III, DNA polymerase β(pol β)]に付加し,ポリADPリボシル化を引き起こす.PARP阻害剤はDNA損傷修復関連タンパク質の集積を抑制するため,DNA修復不全による細胞死を誘導する.
DNA損傷刺激はSTAT1やSTAT3シグナル経路とその下流に存在するinterferon regulatory factor 1(IRF1)を活性化させ,IRF1によってPD-L1の転写が誘導される6).放射線やDNA傷害性抗がん剤の処理によって,STAT1のTyr701とSer727, STAT3のTyr705とSer727がリン酸化されると,これらのリン酸化が引き金となりIRF1を介してPD-L1の転写が誘導される.このことから,DNA損傷下におけるPD-L1の転写はJAK1/2-STAT1/2/3-IRF1経路による制御を受けていることが示唆されている6).乳がんのサブタイプ分類の中でも,トリプルネガティブと呼ばれるホルモン療法や抗HER2療法が効かないタイプのがんではPD-L1の発現上昇がみられており,興味深いことにPARP阻害剤によるGSK3βの不活性化によってPD-L1の転写量が上昇することが報告されている7).このようなPARP阻害剤の添加によるPD-L1の発現上昇は抗PD-L1抗体薬による抗腫瘍効果を向上させると期待されている.GSK3βの不活性化によるPD-L1の転写誘導の分子機構は現在のところ不明である.GSK3βは基質へのリン酸化を介してユビキチン化とそれに続くプロテアソーム系によるタンパク質分解を促す“プライミングキナーゼ”としての機能を有していることが知られており,その代表的な基質が転写因子c-Junとc-Mycである8).c-JunはJunBとともにAP-1複合体を形成する.ホジキンリンパ腫などのがん細胞ではこのAP-1複合体が高活性化状態にあることが知られている.また,c-JunとJunBによるAP-1複合体はPD-L1のエンハンサー領域にあるAP-1応答配列に結合し,PD-L1の転写を誘導する9, 10).同様に,転写因子c-MycもPD-L1遺伝子のプロモーター領域に直接結合することが報告されている11).これらの報告より,GSK3βの不活性化によってc-Junとc-Mycはプロテアソームによる分解を回避し,発現が安定化したc-JunとJunBはPD-L1遺伝子の上流のプロモーター/エンハンサー領域に結合することでPD-L1の転写を誘導している可能性が考えられる.
PARP阻害剤の他にもCHK1阻害剤もPD-L1の転写を上昇させることが報告されている12).CHK1阻害剤によって惹起される細胞質DNAは自然免疫応答経路の一つであるSTING経路を活性化する.STING経路では,まず細胞質に生じた二本鎖DNAによって,サイクリックGMP-AMP合成酵素が活性化し,セカンドメッセンジャーとしてcGAMP(cyclic GMP-AMP)が生成される.STINGへのcGAMPの結合はTBK1によるIRF3のリン酸化を引き起こす.IRF3はp65 NF-κBと複合体を形成すると,PD-L1遺伝子の上流のプロモーター領域に直接結合し,PD-L1の転写を誘導する13).このようにSTING経路はPD-L1の転写誘導の制御機構の一つとして知られている.
2)インターフェロン刺激
インターフェロン(IFN)はウイルス感染を抑制する因子として同定されたサイトカインの一つであり,I型IFN(IFN-α/β)もII型(IFN-γ)もともにPD-L1の転写を誘導する14, 15).IFN-γに関しては,IRF1がPD-L1遺伝子のプロモーター部位に結合し,JAK/STAT経路の活性化によって転写を誘導することがヒト肺がん細胞を用いた解析より明らかとなっている16).悪性黒色腫や腎細胞がん,扁平上皮がんでは,STAT3ではなくSTAT1がIFN-γ刺激によるPD-L1の転写誘導に寄与していると報告されている14).IFN-αに関しては,阻害剤によってp38やSTAT3のリン酸化を抑制すると,IFN-αによって惹起されるPD-L1の転写が抑制されることから,これらの因子が制御に関与していると考えられる15).
IFN-αやIFN-γだけでなく,IL-1α, IL-10, IL-27, IL-32γなどのサイトカインもPD-L1の転写を誘導する14, 17).IL-21の刺激下ではSTAT3ではなくSTAT1が,そしてIL-1αの刺激によってp65 NF-κBがPD-L1の転写を促すことが報告されている14).
3)その他の刺激
オートファジー(autophagy)は非炎症性の細胞死で,栄養ストレス時に細胞内容物を分解する機構として広く知られている.オートファジーはオートファゴソームと呼ばれる二重膜小胞の形成,リソソームとの融合によるオートリソソームの形成,細胞内タンパク質や細胞小器官の分解の3段階のプロセスからなる.興味深いことにオートファジーの誘導はPD-L1の発現を低下させることが報告されている18).ヒト胃がん細胞を用いた解析によるとオートファジー関連遺伝子の一つであるATG5またはATG7のノックダウンはPD-L1の発現を上昇させる.また,リソソーム阻害剤クロロキンやバフィロマイシンA1によってオートファジーの誘導を阻害するとPD-L1の発現の上昇がみられた.そして興味深いことに,その発現上昇はNF-κB経路の活性化と関連性があることが示唆されている18).また,Atg5欠失マウスではTBK1の活性化によるPD-L1の発現上昇が報告されている6).
1)転写制御
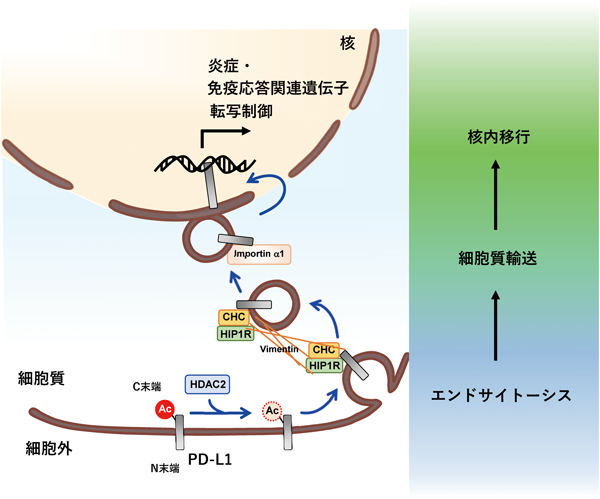
ほとんどのPD-L1は細胞膜上に発現し,PD-1のリガンドとして機能しているが,一方で,ある割合のPD-L1は核内に局在することが2010年に明らかとなっていた5).しかし,核内のPD-L1の機能については長い間不明であった.そこで我々は,核内のPD-L1の機能に着目して研究を進め,PD-L1がDNA上で転写因子p65/RelAやIRFと結合してさまざまな遺伝子の発現制御に寄与していることを見いだした19).RNAシーケンス解析によるとPD-L1ノックアウト細胞では,I型IFNシグナル関連遺伝子やIFN-γシグナル,NF-κBシグナル,MHCクラスI経路などの炎症や免疫応答に関与する遺伝子の発現の低下がみられた(図2)19).このことから,PD-L1の高発現しているがん細胞では炎症や免疫応答に関わる遺伝子群の発現を促すことで,抗PD-1/PD-L1抗体薬に対する感受性を上昇させていると考えられる.事実,hot tumorと呼ばれる免疫系の活性化している腫瘍ではPD-L1の発現が上昇しており,免疫チェックポイント阻害剤が効きやすいという報告もあることから,このようなPD-L1による炎症や免疫応答関連遺伝子群の発現誘導はhot tumorのようながんの微小環境を作り出しているのではないかと考えられる.また,このような免疫応答に関わる遺伝子群の他にも,PD-L1はPD-L2やVISTA, B7-H3のような他のB7ファミリーリガンドの転写誘導にも関わっている19).前述のとおり,これらの免疫チェックポイント分子はT細胞の活性化の抑制に関わっているものもあることから,がん細胞のPD-L1に抗PD-L1抗体薬が作用しても,これらの免疫チェックポイント分子の発現を上昇させることで免疫寛容を引き起こすことが可能となる,つまり抗PD-L1抗体薬への耐性の獲得に寄与しているのではないかと考えられる.我々の報告以外にもオートファジーやmTOR関連遺伝子,また後述するがパイロトーシスと呼ばれる細胞死の誘導に関わる遺伝子の転写誘導に関与していることも報告されている20, 21).
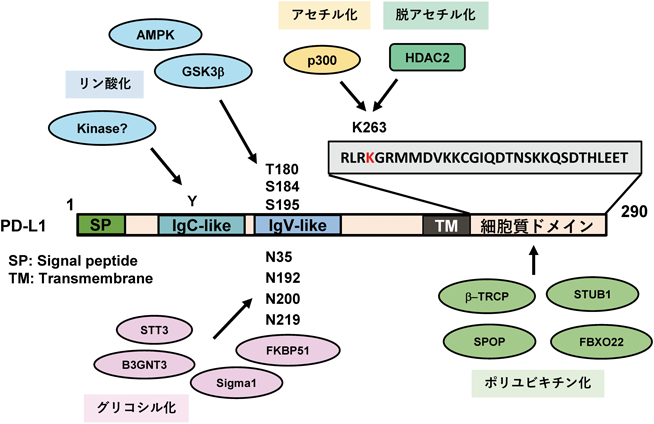
細胞膜上に発現しているPD-L1が核内で機能するためには,核内移行の必要がある.我々はPD-L1の核内移行に着目し,解析を進めた結果,C末端側にある細胞質ドメインの翻訳後修飾が局在制御を担っていることを明らかにした.細胞質ドメイン内のLys263がhistone deacetylase 2(HDAC2)によって脱アセチル化されることで,PD-L1はアダプチンβ2やクラスリン,huntingtin interacting protein 1-related protein(HIP1R),ビメンチン,インポーチンα1との結合が可能となり,細胞膜上からエンドサイトーシス,細胞質輸送,核内移行する19).HDAC2の阻害剤で核内移行を阻害すると,核内での炎症・免疫応答に関わる遺伝子の転写誘導は抑制された.興味深いことに,HDAC2の特異的阻害剤Santacruzamate Aと抗PD-1抗体を,腫瘍を移植したマウスに投与すると全生存率が改善された19).このことから,HDAC2阻害剤はPD-1/PD-L1を標的とした免疫チェックポイント阻害剤との併用療法に有効であることが示唆された.
2)シグナル伝達
a. IFNシグナル
IFN-γなどのIFNはアポトーシスの誘導や細胞増殖の抑制,細胞老化の誘導などがん細胞に対して抗腫瘍的に働く.STAT3は,抗アポトーシス分子であるBcl-2やBcl-xl, Mcl-1の転写を誘導することでがん細胞にアポトーシスを回避させるように働く転写因子である.STAT3のTyr705のリン酸化は二量体形成や核内移行,DNAへの結合を可能とする.PD-L1のノックアウトマウスでは,このSTAT3 Tyr705のリン酸化が上昇していることが明らかとなっている22).さらに,そのノックアウトマウスではIFNを投与した後に,Caspase-7の発現が上昇していた.これらの結果より,PD-L1はIFNによって誘導されるSTAT3やCaspase-7の活性化を抑制していると考えられる.PD-L1の細胞質ドメイン内に存在するRMLDVEKC配列はIFNによる細胞毒性効果に対し抑制的に働くと考えられており,がん患者においてその中に存在する二つのアミノ酸に対する体細胞突然変異(D276HとK280N)が報告されている22).前述のとおり,IFNはPD-L1の発現を増加させる刺激の一つであることから,IFN刺激下のPD-L1によるSTAT3やCaspase-7への制御は負のフィードバック機構であると考えられる.
b. mTORシグナル
mTORはセリン/トレオニンキナーゼで,ラパマイシンの標的分子として発見された.mTORは複数の分子と複合体(mTOR1, mTORC2)を形成し,タンパク質合成や脂質代謝,オートファジーなどfundamentalな生命現象に関与することが広く知られている.そして,興味深いことに,PD-L1の発現の低下はmTORの活性を抑制することが報告されている20, 23).同様に,抗PD-L1抗体薬の添加によってmTORの活性の低下とそれに伴う解糖代謝を抑制することも明らかとなっている24).卵巣がん細胞では,PD-L1の欠失によってmTORC1によるp70S6K Thr389のリン酸化やmTORC2によるAkt Ser473のリン酸化が低下する20).また,PD-L1の機能阻害はラパマイシンの細胞増殖抑制効果を向上させることから,ラパマイシンは抗PD-L1抗体薬との併用療法において抗腫瘍効果を向上させる抗腫瘍薬となると期待される.
3)細胞死
細胞死は形態形成や生体内での恒常性の維持に必須である生命現象の一つであることから,その制御機構の破綻はがんや自己免疫疾患などの発症の起因となっている.細胞死は,制御された細胞死(programmed cell death)とそうでない細胞死(non-programed cell death)の二つのグループに分けられる.制御された細胞死はさらに,炎症性のもの(パイロトーシス)と非炎症性のもの(オートファジー)に分けられる.
a. オートファジー
オートファジーの誘導はPD-L1の発現を低下させる.一方で,PD-L1の欠失はオートファジー誘導関連遺伝子の転写量を上昇させることで,オートファゴソームの形成を促す20).PD-L1の発現の低いがん細胞では,PD-L1を高発現しているがん細胞に比べ,オートファジー阻害剤クロロキンに対する抵抗性が高い20).このことから,がん細胞でのPD-L1の発現はオートファジー阻害剤への応答性に対するバイオマーカーとなると期待される.また,神経膠芽腫の細胞では,PD-L1はAktとともにオートファジー誘導を阻害し,その結果として浸潤能を上昇させる23).神経膠芽腫の組織では,虚血による栄養飢餓とがん細胞の増殖や浸潤には相関があることが報告されている.定常状態の細胞内では,PD-L1の発現はPI3/Akt経路の活性化の指標であるAktのSer473のリン酸化やmTORのSer2448のリン酸化状態に影響は与えないが,細胞を血清飢餓状態で培養するとPD-L1の発現上昇はこれらのリン酸化を亢進させた.オートファジーの誘導はAtg8/LC3の顆粒状(puncta)の集積で検出することができ,血清飢餓状態で培養したPD-L1のノックアウト細胞ではLC3 punctaの形成が促進された.PD-L1はAktと結合することで細胞膜上につなぎ止めPI3/Akt経路を活性化し,さらに細胞の浸潤や細胞骨格系遺伝子の発現を制御することでオートファジーに対して抑制的に働きかけていると考えられている23).前述のとおり,オートファジーの誘導はPD-L1の発現を低下させる18).このことからオートファジーとPD-L1の発現制御は互いに負に制御し合っている可能性がある.
b. パイロトーシス
パイロトーシス(pyroptosis)は,1992年にZychlinskyらによって発見された細胞死の一つで,マクロファージなどの炎症細胞で細菌や病原体の感染によって惹起される現象として発見された.Caspase-1やCaspase-4の活性化と,Caspaseによる切断・Gasdermin Dによって誘導される細胞死である.パイロトーシス誘導におけるPD-L1の機能はあまりよく明らかとなっていないが,興味深いことに,低酸素刺激によってPD-L1は核内にてTyr705がリン酸化されたSTAT3と結合し,Gasdermin Dの発現を誘導することでパイロトーシスを誘導することが報告されている21).このことから,低酸素刺激によって誘導される核内PD-L1によるパイロトーシス誘導はSTAT3依存的であることが示唆されている.
4)mRNAの安定性の維持
前述のとおり,DNA損傷刺激はPD-L1の転写を強く誘導する刺激の一つであるが,発現誘導されたPD-L1が細胞内でどのような機能を担っているかについては長い間不明であった.PD-L1研究を牽引しているグループの一つである米国のMayo Clinicのグループによって,PD-L1がmRNAの安定性に寄与しているとの興味深い研究内容が2019年に報告された.彼らの報告によると,DNA損傷下においてPD-L1はBRCA1やNBS1のmRNAに直接結合し,RNA exosomeによる分解から保護することでmRNAの安定性に関与する25).そして興味深いことに,抗PD-L1抗体の添加はPD-L1の分解とそれに伴うBRCA1やNBS1などのmRNAの分解を引き起こし,DNA損傷に対する感受性を増強させた.
PD-1に対するリガンドとしてのPD-L1の機能については免疫学・腫瘍学によって幅広く研究が進められ,腫瘍形成に対して促進的に働くことが明らかにされてきた.PD-L1の発現の制御に関わる分子は同定されつつあるが(図3),PD-L1の細胞内機能についてはまだ不明な点が多い.本稿で紹介したとおり,PD-L1はさまざまな生物学的にfundamentalな機能を持つことが知られており,細胞膜上のPD-L1と同様に“細胞内のPD-L1もがん化を促進させる分子”と捉えることはできないと著者は考えている.それはPD-L1ががん細胞だけでなく抗原提示細胞などにも発現している分子だからかもしれない.がん治療を目指した,免疫チェックポイント阻害剤との併用療法の確立には,細胞内のPD-L1がどのように腫瘍化に関与しているのかを明らかにする必要がある.そのためにも腫瘍学の観点からのPD-L1のさらなる研究が待たれる.
謝辞Acknowledgments
本稿内で紹介した著者らによる研究内容は,米国Harvard Medical School, Beth Israel Deaconess Medical CenterのWenyi Wei教授と東京医科歯科大学難治疾患研究所分子遺伝分野の三木義男教授の御指導のもと進めてきたものである.研究を進める過程でのディスカッションに時間を割いていただいたこと,実験環境を充実したものに整えていただいたことを心より感謝しております.また,共同研究者であるHarvard Medical School, Dana-Farber Cancer InstituteのPeter Sicinski教授やGordon Freeman教授にも研究実施におけるサポートを賜りましたことを深く感謝いたします.